Atherosclerotic occlusive disease of the cervical and intracranial arteries leads to ischemic stroke through two separate, but interrelated, mechanisms: local thrombosis or embolism from atherosclerotic plaque, and hemodynamic failure (low flow). In this article, the author discusses the evidence linking these two mechanisms with cerebral ischemia, and the evidence for the synergistic effects of thromboembolism and impaired hemodynamics. An understanding of these two mechanisms is important because these mechanisms provide the rationale for revascularization for patients who have atherosclerotic stenosis or occlusion. In addition, the biologic imaging of atherosclerotic plaques and hemodynamic assessment eventually will play an important role in stratifying patient risk and guiding physiologically based patient selection for intervention.
Atherosclerotic disease of the cervical and intracranial arteries is a frequent cause of stroke. This causality has been established by the frequent association of large artery atherosclerotic (LAA) disease and ischemic events and, more importantly, by the benefit of surgical revascularization in large, randomized trials of carotid endarterectomy for patients who had symptomatic extracranial carotid stenosis and some subgroups of patients who had asymptomatic stenosis . Likely, both thromboembolic and hemodynamic mechanisms are involved in the pathogenesis of stroke in these patients. In this article, the author reviews the data supporting thromboembolic, hemodynamic, and synergistic mechanisms. An understanding of these mechanisms is important because it is likely that the imaging methods of establishing thromboembolic or hemodynamic risk will become important in identifying patients for intervention, possibly including patients who have asymptomatic atherosclerotic lesions and who are at high risk for future stroke.
The frequency of large artery extracranial or intracranial atherosclerotic disease in patients presenting with transient ischemic attack or stroke varies from study to study, likely owing to racial or environmental factors and study methodology (ie, method of anatomic imaging, if any). Extracranial carotid stenosis or occlusion is among the most common lesions identified in patients presenting with transient ischemic attack or stroke. In a population-based study in Manchester, England, severe carotid stenosis or occlusion was found ipsilateral to the ischemic event in nearly 25% of patients presenting with stroke during a 1-year period . A similar frequency of carotid disease was found in a large series of patients presenting with transient ischemic attack . Intracranial and vertebral-origin lesions are less frequent, accounting for 5% to 10% of patients presenting with stroke . Small-vessel arterial disease and cardiac conditions, such as atrial fibrillation, also account for a large proportion of ischemic stroke.
Cerebral ischemia
Brain tissue requires the continuous delivery of oxygen and glucose to maintain normal neuronal function. When an artery becomes occluded, the brain supplied by that artery may become ischemic, depending on the adequacy of flow through collateral channels. The cortical surface of the brain often, but not always, has an abundant network of pial collaterals connecting arterial branches. Penetrating arteries into the white matter do not have the benefit of such a network . The survival of brain cells during a period of ischemia is related to several factors. When the delivery of blood to a region of brain tissue falls below a critical threshold, 30 mL of blood per 100 g of tissue per minute in some studies, normal neuronal function ceases. This cessation is related to the reduced delivery of oxygen, not glucose. Glucose is brought into the cells by an active transport mechanism. If flow is restored rapidly, no permanent injury may occur. Irreversible ischemic injury critically depends on both the depth and duration of ischemia . Other factors are also important, including brain temperature, serum glucose levels, and the area of the brain affected. The differential effects on the different components of the neurovascular unit (ie, the neuron, endothelium, and glia) are understood poorly . A thromboembolic stroke often results in a great deal of regional variability in the depth of ischemia. Some ischemic areas may remain viable for up to 24 hours . These regions of ischemic, but viable, tissue have been termed the “ischemic penumbra” . The improved outcome of patients who have been revascularized in the hours after stroke onset has validated this method empirically .
Compensatory mechanisms to low flow
Patients who have symptomatic atherosclerotic occlusive disease may have reduced flow to the distal territory, owing to poor sources of collateral flow. Conversely, many patients who have occlusive disease, even complete occlusion, have normal flow, owing to good collateral sources . When perfusion pressure (the difference between mean arterial pressure and the venous backpressure) falls in any arterial territory, the brain and brain vasculature may compensate through two mechanisms ( Fig. 1 ) . The first mechanism is arterial dilation and the second is an increase in the amount of oxygen removed from the blood. Autoregulatory vasodilation is a response by penetrating capacitance arterioles to either reductions in arterial pressure or reduced arterial oxygen content . Neuronal, muscular, and physiologic factors are involved in this complex reflex. Arterial resistance is reduced, allowing flow to continue at near-normal rates over a wide range of pressures . Some slight reduction in flow occurs through the autoregulatory range . At some threshold point, however, further reductions in pressure exceed the ability of vasodilation to compensate, and flow falls passively as a function of pressure. This situation is known as autoregulatory failure.
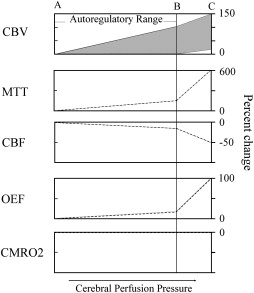
The compensatory reflex to reduced blood flow is an increase in tissue oxygen extraction . For a given tissue metabolic rate for oxygen consumption, a reduction in the delivery of oxygen (cerebral blood flow) requires an increase in the amount of oxygen extracted by the blood (oxygen extraction fraction [OEF]) . Normal OEF is 30% to 40%, and can increase instantaneously to up to 80%. The precise mechanism by which this increase occurs is not clear. Oxygen is not transported, and reaches the brain tissue by passive diffusion. Tissue oxygen tensions are not reduced when flow falls. It is likely that OEF increases because of a constant flux of oxygen between the tissue and the blood. When flow falls, there is a relative increase in the amount of oxygen used by the brain .
In humans, these compensatory mechanisms are identified by several different physiologic imaging methods . Anatomic imaging studies are not sufficient to determine the hemodynamic status in individual patients . Clinical symptoms, with the exception of orthostatic or hemodynamic transient ischemic attacks , also are not predictive of hemodynamic status. As a consequence, three broad categories of imaging methods have been developed. Paired flow tests assess the ability of the cerebrovasculature to increase blood flow or blood velocity to a vasodilatory stimulus. If flow fails to increase, pre-existing autoregulatory vasodilation is inferred. Static measurements of cerebral blood flow, blood volume, and mean transit time are another method for identifying the presence of autoregulatory vasodilation. Finally, OEF may be measured directly ( Fig. 2 ). These methods are not interchangeable; different physiologic mechanisms are being assessed. The association between stroke risk and these methods, or the correlation between a candidate method and a proven method, must be proven.
Compensatory mechanisms to low flow
Patients who have symptomatic atherosclerotic occlusive disease may have reduced flow to the distal territory, owing to poor sources of collateral flow. Conversely, many patients who have occlusive disease, even complete occlusion, have normal flow, owing to good collateral sources . When perfusion pressure (the difference between mean arterial pressure and the venous backpressure) falls in any arterial territory, the brain and brain vasculature may compensate through two mechanisms ( Fig. 1 ) . The first mechanism is arterial dilation and the second is an increase in the amount of oxygen removed from the blood. Autoregulatory vasodilation is a response by penetrating capacitance arterioles to either reductions in arterial pressure or reduced arterial oxygen content . Neuronal, muscular, and physiologic factors are involved in this complex reflex. Arterial resistance is reduced, allowing flow to continue at near-normal rates over a wide range of pressures . Some slight reduction in flow occurs through the autoregulatory range . At some threshold point, however, further reductions in pressure exceed the ability of vasodilation to compensate, and flow falls passively as a function of pressure. This situation is known as autoregulatory failure.
The compensatory reflex to reduced blood flow is an increase in tissue oxygen extraction . For a given tissue metabolic rate for oxygen consumption, a reduction in the delivery of oxygen (cerebral blood flow) requires an increase in the amount of oxygen extracted by the blood (oxygen extraction fraction [OEF]) . Normal OEF is 30% to 40%, and can increase instantaneously to up to 80%. The precise mechanism by which this increase occurs is not clear. Oxygen is not transported, and reaches the brain tissue by passive diffusion. Tissue oxygen tensions are not reduced when flow falls. It is likely that OEF increases because of a constant flux of oxygen between the tissue and the blood. When flow falls, there is a relative increase in the amount of oxygen used by the brain .
In humans, these compensatory mechanisms are identified by several different physiologic imaging methods . Anatomic imaging studies are not sufficient to determine the hemodynamic status in individual patients . Clinical symptoms, with the exception of orthostatic or hemodynamic transient ischemic attacks , also are not predictive of hemodynamic status. As a consequence, three broad categories of imaging methods have been developed. Paired flow tests assess the ability of the cerebrovasculature to increase blood flow or blood velocity to a vasodilatory stimulus. If flow fails to increase, pre-existing autoregulatory vasodilation is inferred. Static measurements of cerebral blood flow, blood volume, and mean transit time are another method for identifying the presence of autoregulatory vasodilation. Finally, OEF may be measured directly ( Fig. 2 ). These methods are not interchangeable; different physiologic mechanisms are being assessed. The association between stroke risk and these methods, or the correlation between a candidate method and a proven method, must be proven.
Thromboembolic mechanisms
Thromboembolism from atherosclerotic plaque is the primary mechanism of stroke in most patients who have LAA. The current concept of the vulnerable plaque is accepted widely and is discussed fully elsewhere in this issue. Plaque rupture may lead to platelet aggregation, local thrombosis, or thromboembolism. Plaque material, itself, may embolize .
Most of the current knowledge regarding the biology of atherosclerotic plaque and the cascade of events that lead to plaque rupture and platelet aggregation has come from human coronary artery studies and animal models of atherosclerosis. It is likely that these same mechanisms are involved in cerebrovascular atherosclerotic disease. Unlike coronary circulation, local thrombosis is a less common presentation than distal embolism. However, two key differences between coronary and cerebral circulation may make the coronary circulation more vulnerable to local thrombosis and occlusion at the site of the atherosclerotic lesion. The first difference is the continuous high flow rate in cerebral circulation. Diastolic flow rates are generally 40% of peak systolic flow. During heart contraction (systole), there is no antegrade coronary artery flow. This episodic stasis may make local thrombotic occlusion more likely to occur. The second factor that may predispose to embolism rather than local occlusion of cerebrovascular vessels is the relatively large vessel diameters of the extracranial vessels, relative to the coronary arteries.
The clinical evidence linking local stenosis to distal embolism is very strong. Platelet thrombi have been observed in retinal vessels distal to atherosclerotic stenoses and have also been identified on surgical specimens after carotid endarterectomy. Single or multiple distal emboli often are identified on angiography in patients who have atherosclerotic carotid disease after acute ischemic stroke . Diffusion-weighted imaging in these patients most often shows single or multiple mixed cortical and white matter ischemic lesions, suggesting emboli, even in patients who have clinical lacunar syndromes . Finally, embolic events result commonly from symptomatic carotid stenosis. Molloy and Markus monitored the middle cerebral artery for embolic signals using transcranial Doppler (TCD) in patients who had had a recent transient ischemic attack or stroke and severe stenosis of the ipsilateral extracranial carotid artery. Frequent transient high-intensity signals were found in patients who had symptomatic carotid stenosis. No signals were observed in a group of asymptomatic patients who had similar degrees of stenosis.
Related posts:
 Pathology of Atherosclerosis and Stenting
Pathology of Atherosclerosis and Stenting
 Extracranial Stenosis: Endovascular Treatment
Technique for Intracranial Balloon and Stent-Assisted Angioplasty for Atherosclerotic Stenosis
Angioplasty and Stenting for Atherosclerotic Intracranial Stenosis: Rationale for a Randomized Clinical Trial
Endovascular Treatment of Vertebral Artery–Origin and Innominate/Subclavian Disease: Indications and Technique
Extracranial Stenosis: Endovascular Treatment
Technique for Intracranial Balloon and Stent-Assisted Angioplasty for Atherosclerotic Stenosis
Angioplasty and Stenting for Atherosclerotic Intracranial Stenosis: Rationale for a Randomized Clinical Trial
Endovascular Treatment of Vertebral Artery–Origin and Innominate/Subclavian Disease: Indications and Technique
 Techniques of Carotid Angioplasty and Stenting
Techniques of Carotid Angioplasty and Stenting
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


