Fig. 1
Simplified schematic diagram of the molecular events underlying development of normal tissue toxicity following radiation exposure (Bentzen 2006)
At the time of irradiation, the production of hydroxyl radical due to radiolytic hydrolysis of water can lead to single or double stranded breaks within the DNA. Single stranded breaks are more easily repaired and rarely cause lethal injury to the cell where as double stranded breaks often cause cell death either due to apoptosis or mitotic catastrophe. The classical, “target cell theory” of radiation-injury suggested that organ-specific sequelae are a result of a threshold level of cell death within a target cell population. The target cell theory suggested that the so-called “latent period” between the time of exposure and the development of symptomatic injury was the direct result of the proliferation kinetics of a critical cell population required for proper tissue function. Based on this, the timing and expression of injury could be considered a result of the proliferation kinetics of the irradiated tissue (Michalowski 1986). Thus, organs with a rapidly renewing stem cell compartments (i.e., gastrointestinal tissue) would develop injury earlier than tissues characterized by well-differentiated cell populations (i.e., lung).
How cells recognize and respond to injury may influence the tissue response to radiation (McBride et al. 2004) Although only a few cells may be directly targeted by the ionizing event, bystander effects on non-irradiated cells have been observed up to 1 mm away by Brenner and colleagues (Belyakov et al. 2005). In the lung, as well as other “late responding tissues”, the relevance of the target cell theory has been challenged in recent years. Based on work performed over the past several decades implicating a wide range of dynamic signaling events determining whether a cell lives or dies and communication of the trauma to surrounding tissues, it is becoming clear that the tissue response to radiation is driven by a dynamic tissue response that begins at the time of irradiation and continues throughout the course of disease progression.
3 Role of Free Radical Species and Redox Signaling in Radiation Response
Ionizing radiation directly damages DNA leading to genomic instability and cell death through apoptosis or mitotic catastrophe (O’Neill and Wardman 2009). Investigators evaluating radiation effects on DNA have found that high linear energy transfer results in “clustered DNA lesions” which are inefficiently repaired and are thus thought to be biologically relevant to the acute and delayed effects associated with radiation exposure (O’Neill and Wardman 2009). The ionizing event also results in formation of reactive oxygen species (ROS), however the levels generated by therapeutic doses of radiation are negligible compared to those generated by aerobic metabolism (Ward 1994). Within hours of the initial ionizing exposure, there is a robust activation of secondary sources of oxidative stress leading to persistent production of free radical species that can participate in cell signaling. A number of transcription factors and pro-inflammatory/pro-fibrogenic molecules, such as NFκB, HIF-1α, and TGF-β, are sensitive to changes in the redox status of the cell. As the level of free radical production is thought to be beyond the capacity of the cell’s antioxidant defense mechanisms to eliminate them, the imbalance between free radicals and antioxidants leads to chronic upregulation of transcription factors, growth factors, and cytokines involved in radiation injury (Robbins and Zhao 2004; Benderitter et al. 2007; Zhao et al. 2007). The environment of chronic oxidative stress results in oxidative damage to DNA leading to chromatin remodeling and altered gene expression, chronic protein activation/inactivation, and oxidation/peroxidation of membrane lipids that can result in changes in cell signaling (Mikkelsen and Wardman 2003). The ability of potent superoxide dismutase (SOD)-like compounds and other antioxidants to mitigate radiation-induced injury is further evidence that redox-regulated signaling plays a substantial role in the development of radiation injury (Epperly et al. 1999a, b, 2002; Gauter-Fleckenstein et al. 2007, 2010; Rabbani et al. 2005, 2007; Vujaskovic et al. 2002; Rosenthal et al. 2011).
While the exact mechanism through which radiation-induced free radical production affects the activity of signal transduction pathways and transcription factors within the irradiated tissue is not clear, it is well known that many signaling mechanisms and gene expression pathways are sensitive to changes in intracellular oxidation/reduction status (Mikkelsen and Wardman 2003) leading to changes in signal transduction, transcription factor activation, and gene expression (Mikkelsen and Wardman 2003; Reboucas 2010; Zhang and Hogg 2005). It is theorized that the increase in ROS/RNS production in the early radiation response may result in oxidative modification of macromolecules participating in signal transduction pathways, resulting in signaling changes that perpetuate tissue injury over time (Reboucas 2010).
4 Biological Sources of ROS/RNS and the Role of Oxidative Stress in Radiation-Induced Tissue Injury
It is hypothesized that the initial ionizing event is followed by a ROS/RNS mediated signaling cascade through downstream activation of metabolic sources of prooxidant production. This includes mitochondria, nitric oxide synthases, and oxidoreductase enzymes, most notably the family of NADPH oxidases.
Non-phagocytic cells predominantly generate ROS/RNS through activity of the cell membrane-associated family of NADPH oxidases (von Lohneysen et al. 2010). A number of NADPH oxidase family members exist and have remarkably varied activation mechanisms and tissue distribution. For example, endothelial cells express Nox1, Nox2 (gp91phox), Nox4, and Nox5 isoforms; whereas vascular smooth muscle cells express the Nox1, Nox4, and Nox5 isoforms (Lassegue and Griendling 2010). NADPH oxidase-derived superoxide anion (O2. –) is involved in vascular maintenance and regulation of vascular smooth muscle tone under physiological conditions (Bengtsson et al. 2003). However, pathological overproduction of O2. − by NADPH oxidases may participate in radiation injury by activating redox-sensitive signaling pathways involved in inflammation and fibrosis. Collins-Underwood et al. (2008) found that increased expression of NADPH oxidase subunits (Nox4, p22phox, and p47phox) following in vitro irradiation of rat brain microvascular endothelial cells was associated with a pro-inflammatory response. In these studies, elevated intracellular generation of ROS, activation of the pro-inflammatory transcription factor NFκB, and expression of vascular adhesion molecules ICAM-1 and PAI-1 could be prevented by inhibition of NADPH oxidase (Collins-Underwood et al. 2008).
Nox4 is especially interesting as recent studies have shown TGF-β1 induced cell death to be dependent upon Nox4-derived H2O2 (Carnesecchi et al. 2011). Nox4 is a unique isoform as it produces H2O2 rather than O2. − due to an extra 23-amino acid sequence in its E-loop (Takac et al. 2011). In recent studies by Zhang et al. (2012), Nox4 expression was colocalized with increased PTEN expression, which correlated to a progressive and persistent increase in cell death between 24 h and 6 months after whole thorax irradiation. The increase in PTEN expression coincided with downregulation of Akt signaling.
4.1 Amplification of Existing ROS/RNS
Production of ROS/RNS by NADPH oxidases or dysfunctional mitochondria can be amplified by the reaction of nitric oxide and superoxide anion to form peroxynitrite (ONOO−), a potent free radical species that has been implicated in proteoglycan degradation and matrix turnover (Cernanec et al. 2007). Depletion of NO through its rapid conversion to ONOO− or superoxide mediated inactivation of key components of the NO signaling pathway may also contribute to vasoconstriction observed after radiation (Ward et al. 1983; Fleckenstein et al. 2007).
4.2 Factors Affecting ROS/RNS Production by Theses Sources
It is well known that TGF-β, a ubiquitous cytokine activated by radiation, and angiotensin II exert upstream influence over NADPH oxidase activation. The use of angiotensin converting enzyme (ACE) inhibitors to mitigate radiation pneumonitis/fibrosis and nephropathy has been an active area of research primarily conducted by Moulder and colleagues (Ghosh et al. 2009). Pre-clinical studies using a rat model have shown that clinical doses of Captopril (34 mg/kg/day) improve survival and restore vascular reactivity in irradiated lung (Ghosh et al. 2009). The equally protective ability of both thiol and non-thiol containing ACE inhibitors suggests that free radical scavenging is not the primary mechanism of action for mitigation of radiation toxicity. Possible mechanisms include inhibition of AngII-mediated activation of NADPH oxidase and/or effects on blood pressure (Moulder et al. 2002). The role of blood pressure in reducing radiation-induced injury is supported by preclinical studies showing that a diet high in sodium ameliorates radiation nephropathy ostensibly through inhibition of ACE activity (Moulder et al. 2002). The reduction in nephropathy was only observed when a high sodium diet was initiated before the onset of nephropathy; it was not an effective treatment of existing nephropathy (Zhang and Rabbani 2012).
It is of note that the protective role of ACE inhibitors in preclinical models has, to date, not translated to the clinic. Wang and Anscher performed a retrospective study to determine the development and onset of radiation pneumonitis, determined by the NCI common toxicity criteria and CT imaging, in 213 patients treated for lung cancer with thoracic irradiation from 1994 to 1997 (Wang et al. 2000). In this study, the authors found the incidence and onset of radiation pneumonitis among patients taking ACE inhibitors (captopril, benazepril, enalapril, lisinopril, quinapril, or fosinopril) for hypertension was not significantly different than those who were not on ACE inhibitors (Wang et al. 2000). Thus, it remains unclear whether the mitigating effect of ACE inhibitors in preclinical models will translate to the clinic.
5 The “Perpetual Cytokine Cascade” After Radiation Exposure
Within the first week following exposure to radiation, there is a decrease in inflammatory cells followed by an increase in neutrophils and lymphocyte accumulation that quickly returns to normal. Over time, fluctuations in inflammatory cell subsets infiltrating the irradiated tissue can be observed. The “perpetual cascade of cytokines” in the lungs after thoracic irradiation was first proposed by Rubin et al. 1995 which demonstrated an absence of a “latent period” at the molecular and cellular level in late responding tissues.
Our group has helped to pioneer the use of increasingly sophisticated molecular biologic methods to assess and analyze late effects in normal tissue (LENT). In so doing, we have extended the paradigm o the clinical pathologic course of events following irradiation or chemotherapy from target cells per se to include and emphasize the intercellular communication. In our current paradigm, during the course of LENT induction, the so-called cytokine cascade can be considered to involve four basic cell components in tissues or organs—the parenchymal cell, the inflammatory cell, the endothelial cell, and the interstitial cell. These are illustrated in Fig. 2a, are a few of the proinflammatory and profibrotic cytokines that are thought to be expressed concurrently (Rubin and Williams 2001).
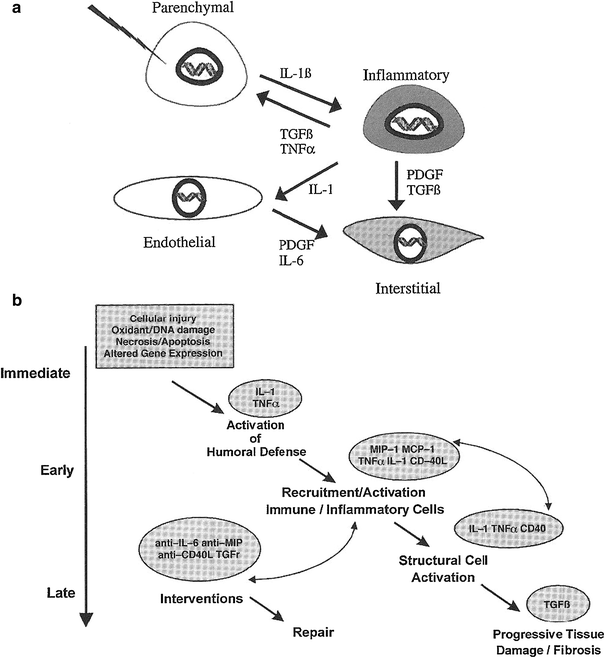
Fig. 2
a Suggested chain of events beginning with the initial injury to the primary target cell—the parenchymal cell—and culminating in activation of the interstitial cells (e.g., fibroblasts) to lay down extracellular matrix. b Hypothetical pathway indicating the chain of events from initial injury to the final late effect (e.g., fibrosis) (With permission from Rubin and Williams 2001)
An intercellular conversation is initiated at the moment of irradiation, when injury to cell components occurs (e.g., membrane, cytoplasmic body, or DNA), which leads to altered gene expression. This reaction is often in the form of an immediate release of cytokine mRNA; in time, this reaction may provoke a series of downstream events through cell signaling, which is illustrated in Fig. 2b. Through signal transduction, the receptor cells are activated; such activation may result in as little as additional or sequential cytokine expression or it can lead to proliferation or production of extracellular matrix proteins, depending on the species of receptor cell. In the specific case of the receptor cell being a fibroblast, activation of the collagen gene, which has been seen within 24 h of injury, can persist for days, weeks, or even months. This extended persistence may continue until the pathologic or clinical expression of injury is manifested (Rubin and Williams 2001).
Using a mixture of molecular biologic techniques and in vivo/in vitro assays, a number of in-field effects can be appreciated. The release of cytokines occurs shortly after irradiation and persists until the pathologic and clinical expression of late effects, and there is an arbitrary temporal division of cytokine expression:
-
Immediate: first 24 h after injury
-
Early: days to ~8 weeks post injury
-
Late: 3–6 months post injury.
This scenario can be seen to correlate and give further credence to the postulated shape of the clinical pathologic course of events. The potential to use cytokines to alter the therapeutic ratio favorably is of great interest to clinicians in the protocol design of new clinical trials.
The most predominant cytokines involved in inflammation appear to be IL-1α/β, IL-6, TNF-α, and TGF-β (Rubin et al. 1995; Chiang et al. 1997; Herskind et al. 1998; Rube et al. 2004; Brush et al. 2007), however the pattern and timing of cytokine expression appears to vary among studies (Hill 2005), as well as among animal models. The variation of cytokine expression in response to radiation between mouse strains may present one possible mechanism for the observed difference in strain sensitivity and response to radiation. Likewise, baseline differences in the number and types of resident inflammatory cell types among strains as well as genetic differences in leukocyte recruitment may contribute to the variation in radiation response. For example, fibrosing strains have a higher ratio of active to latent TGF-β than non-fibrosing strains (Franko et al. 1997). Epperly et al. (1999) found a time dependent progression in pathological fibrosis coincided with increased IL-1 mRNA levels and a biphasic wave of TGF-β expression during the development of fibrotic disease in C57BL/6 J mice (Epperly et al. 1999). However, targeting cytokine production has not been completely effective in improving survival and respiratory function, nor has any single circulating cytokine been identified as a potential biomarker for susceptibility to development of radiation-induced injury.
Cytokines expressed in irradiated tissue, such as interleukins, monocyte chemoattractant protein-1, and keratinocyte chemoattractant (KC), are consistent with what one would expect based on the inflammatory cell infiltrates found in the tissue and in circulating plasma. Inflammatory cells accumulate in irradiated tissue due to persistent upregulation of vascular adhesion markers (Epperly et al. 2002; Son et al. 2006; Muller et al. 2006; Jaal and Dorr 2005; Ikeda et al. 2000; Olschowka et al. 1997; Gaugler et al. 1997). Targeting inflammatory cell recruitment through inhibition of vascular adhesion molecules, specifically ICAM-1, reduces the number of infiltrating inflammatory cells and blocks the development of fibrosis in irradiated lungs of C57BL/6 mice (Hallahan et al. 2002; Hallahan and Virudachalam 1997). Although the impact of systemically blocking ICAM-1 on acute and chronic inflammation is significant, there is no improvement in respiratory function or survival among non-treated and treated animals exposed to radiation (Hallahan et al. 2002). This suggests an underlying cause for radiation pneumonitis/fibrosis, which impacts animal survival, distinct from inflammation alone.
5.1 Transforming Growth Factor-Beta 1
TGF-β1 is a multipotent cytokine that activates transcriptional regulators and signal transducers involved in cellular proliferation, immunosuppression, extracellular matrix remodeling and inhibition of matrix degradation, chronic inflammatory disease, and angiogenesis (Flanders 2004; Lewis et al. 1999). TGF-β is secreted as an inactive polypeptide by virtually all cells, including fibroblasts, epithelial cells, macrophages, and others. Free radicals produced during radiation can activate TGF-β, which can then bind the TGF-β type II receptor (Ehrhart et al. 1997) setting off a phosphorylation cascade that transduces the signal from the cell membrane to the nucleus (Flanders 2004; Roberts 1999). Interestingly, it has recently been shown that the Smad2/3 phosphorylation cascade downstream of TGF-β1 is dependent on Nox4 signaling further suggesting a critical role of redox signaling in propagating fibrosis (Amara et al. 2010).
Franko and colleagues 1997 found differential expression of TGF-β latency associated peptide (LAP) in the lungs of C57LJ and C3HeB/FeJ mice after irradiation (11 and 18 Gy respectively). In the C57L/J mice, a significantly greater proportion of macrophages and uninvolved tissue expressed TGF-β LAP as compared to lesions with acute/intense inflammation and fibrosis. Cells positive for LAP within the areas of intense inflammation were predominantly macrophages and Type II Pneumocytes. This was in contrast to C3HeB/FeJ, a non-fibrosing mouse strain, which showed significantly higher levels of latent TGF-β (Franko et al. 1997). Since TGF-β is secreted as an inactive molecule bound to the extracellular matrix by its LAP, the results of Franko suggest Type II pneumocytes and macrophages are the predominant producers of TGF-β following radiation in fibrosing mouse strains (C57L background).
Studies by Epperly et al. (1999) found decreased early onset of TGF-β, IL-1, and TNF-α mRNA levels in irradiated lung following intratracheal administration of a manganese-SOD plasmid/liposome complex correlated with improved median survival time. It is important to note that at the time of death, TGF-β levels were not significantly different among groups. Several studies using pharmacologic inhibitors to TGF-β or components of its signal transduction pathway have the ability to significantly reduce radiation fibrosis and improve lung function, indicating that unregulated TGF-β is a mediator of fibroproliferative disease (Flanders 2004; Anscher et al. 2008; Biswas et al. 2007; Nishioka et al. 2004; Xavier et al. 2004).
6 Vascular Dysfunction Following Ionizing Radiation
It has long been acknowledged that the vascular endothelium may be an important target of ionizing radiation. Indeed, several studies have shown both gastrointestinal toxicity and xerostomia to be influenced by endothelial cell damage. Ionizing radiation can disrupt the structural integrity of the vascular architecture resulting in endothelial cell damage, barrier breakdown, leakage, and edema (Anscher et al. 2005; Martin et al. 2000; Stone et al. 2004). As a consequence of the disruption in vascular function, hypoxic regions can develop in the under-perfused tissue. Indeed, one of the key pathophysiological consequences of vascular injury after radiation is the development of tissue hypoxia. Tissue hypoxia can recruit inflammatory cells, where they are then activated to undergo the respiratory burst. Not only does this increase oxidative stress, but also further contributes to tissue hypoxia as oxygen is required for the respiratory burst in phagocytic cells. One of the primary cell types recruited by hypoxia is the macrophage. Jackson et al., demonstrated that macrophages incubated under hypoxic conditions exhibit a redox-dependent increase in TGF-β production and the hypoxia-inducible factor-1 alpha (HIF-1α) product VEGF (Jackson et al. 2007).
HIF-1α has observed in lung tissue as early as one day after radiation. The ability of free radical scavenging to inhibit stabilization of HIF-1α has implicated ROS/RNS as a critical activator of HIF transcriptional activity (Pouyssegur and Mechta-Grigoriou 2006; Li et al. 2007). The consequences of HIF stabilization in irradiated healthy tissue likely include greater endothelial dysfunction, enhanced vascular hyperpermeability, formation of temporary fibrin matrices, and angiogenesis (van Hinsbergh et al. 2001; van Hinsbergh 2001a, b; Semenza 2000). Vascular leakage and fibrin deposition facilitate the formation of new vessels (van Hinsbergh et al. 2001) through establishment of a provisional fibrin matrix which provides a stable scaffold for HIF-1α mediated inflammatory and endothelial cell migration and vessel sprouting (Haroon et al. 1999). Under physiological conditions, HIF has a relatively short half-life of less than 5 min (Wang et al. 1995; Huang et al. 1998). However, under hypoxia, or in the event of oxidative/nitroxidative modification of protein residues in the oxygen dependent domain, the alpha subunit of HIF is stabilized and translocated to the nucleus where it forms a heterodimer with its constitutively expressed HIF-1β subunit. The HIF heterodimer binds to the hypoxia response element (HRE) of a wide variety of genes regulating cell proliferation, migration, pH, apoptosis, energy metabolism, and most importantly, angiogenesis (Brahimi-Horn et al. 2005).
Vujaskovic et al. showed an increase in expression of the HIF product vascular endothelial growth factor (VEGF) between the time of radiation exposure and development of symptomatic disease in a rat model of radiation-induced lung injury (Fleckenstein et al. 2007
Related posts:
 and Biointervention: Cytokine Pathways as a Rationale for Anti-cytokine Interventions Post-Radiation
and Biointervention: Cytokine Pathways as a Rationale for Anti-cytokine Interventions Post-Radiation
 Bioepidemiology of Multiple Primary Cancers
Consequences of Late Effects
Bioepidemiology of Multiple Primary Cancers
Consequences of Late Effects
 of the Pathophysiology Paradigm
of the Pathophysiology Paradigm
 of Normal Tissue TNM Toxicity Taxonomy: Scoring the Adverse Effects of Cancer Treatment
of Normal Tissue TNM Toxicity Taxonomy: Scoring the Adverse Effects of Cancer Treatment
 Analyses of RT-Induced Late Normal Tissue Injury Using Functional Imaging
Analyses of RT-Induced Late Normal Tissue Injury Using Functional Imaging
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


