Etiology
Mixed connective tissue disease (MCTD) is a disease with certain features of polymyositis, scleroderma, and systemic lupus erythematosus. Much of the evidence that MCTD is a distinct clinical entity stems from the identification of antiribonucleoprotein (anti-RNP) antibody, disease-specific human leukocyte antigen (HLA) profiles, suggestive clinical features, and because in the vast majority of patients, MCTD does not evolve into other connective tissue disease–related entities.
The etiology of MCTD is unknown, but the presence of antibodies to RNPs implies that the immune system has become sensitized to ribonucleic acid synthesis or metabolism. Infectious agents have been investigated as a potential trigger for MCTD. Retroviral sequences have been found in a high proportion of MCTD patients, and most studies have revealed a strong association with MCTD and HLA-DR4.
Prevalence and Epidemiology
The incidence of MCTD is not known but is estimated to be higher than that of polymyositis, lower than that of systemic lupus erythematosus, and similar to that of systemic sclerosis. The majority of patients are women, with an average age at diagnosis of 37 years (range, 4–80 years). Pediatric-onset MCTD carries less mortality than adult-onset MCTD, probably because pulmonary hypertension is less frequent in the younger group. The prognosis for patients with MCTD is highly variable, but a severe, progressive disease course is unusual. In five series of MCTD (total of 194 patients), mortality was generally low (mean, 13%; range, 4%–35%); nevertheless, the fact that MCTD affects young women indicates a greater risk for premature death. Pulmonary hypertension is the most serious complication of MCTD, with severe infection secondary to immunosuppression being the other frequent cause of death.
Clinical Presentation
Most patients with MCTD have one or more of the following: Raynaud phenomenon, arthralgia, arthritis, swollen hands, sclerodactyly, or acrosclerosis (i.e., stiffness and atrophy of the skin of hands and feet), and two-thirds suffer from frank myositis. Esophageal dysmotility and reflux are reportedly frequent in MCTD, and generalized lymphadenopathy has been observed in 50% of patients. The most common cardiac abnormality is pericarditis; less common cardiac manifestations include myocarditis and complete heart block. The most frequent clinical manifestations of pulmonary involvement are dyspnea, pleuritic chest pain, and bibasilar rales. Secondary Sjögren syndrome is relatively common. Cutaneous manifestations include photosensitivity, livedo reticularis (a net-like vascular skin discoloration in the trunk or extremities), and calcinosis.
When Sharp and colleagues first described MCTD, it was considered a benign condition requiring little treatment. It is now realized that serious renal and pulmonary involvement does occur; interstitial lung disease occurs in 21% to 66% of patients.
Pleural effusion is one of the most common clinical manifestations of MCTD, but it is usually small and resolves spontaneously. In a retrospective review of the chest radiographs of 81 patients with MCTD, 6% had small pleural effusions, and even fewer had pleural thickening. Although a prospective study of patients with “MCTD” reported “pleuritis” in 35%, this study was undertaken before modern diagnostic criteria had been published. The incidence of pleuritis and effusions is likely to fall in the middle of this range because chest radiography may underestimate disease (compared with computed tomography [CT]) and, conversely, modern diagnostic criteria for MCTD are more rigorous.
MCTD is associated with pulmonary hypertension, commonly manifested insidiously as progressive dyspnea on exertion. A surveillance study of a drug (bosentan) used to treat pulmonary hypertension found that connective tissue disease–related pulmonary hypertension and idiopathic pulmonary hypertension were each responsible for approximately a third of cases treated with this drug. In this cohort the majority of patients with connective tissue disease–associated pulmonary hypertension had systemic sclerosis (75%), and the next largest group consisted of patients with MCTD (9%). Although having a lower incidence in MCTD than in systemic sclerosis, pulmonary hypertension should be considered as a possible cause of dyspnea in both diseases.
Pathophysiology
Pathology
In patients with pulmonary hypertension in MCTD, plexiform lesions in small- and medium-sized arteries can occur, similar to primary pulmonary hypertension. In other connective tissue diseases, pulmonary vasculopathies are often associated with diffuse alveolar hemorrhage, but reports of diffuse alveolar hemorrhage in patients with MCTD are rare. Pulmonary thromboembolism may occur, possibly secondary to coexisting antiphospholipid syndrome.
Pulmonary hemorrhage, diffuse alveolar damage, organizing pneumonia, nonspecific interstitial pneumonia, usual interstitial pneumonia (UIP), and airway disease may be seen in MCTD. These findings are consistent with the clinical manifestations of MCTD, which commonly have features of systemic sclerosis, systemic lupus erythematosus, or polymyositis/dermatomyositis.
Lung Function
Pulmonary disease has been reported in 73% of patients with MCTD on the basis of pulmonary function tests, chest radiography, or both. The most common abnormality in pulmonary function testing is decreased lung diffusing capacity for carbon monoxide (DLCO) ; nearly half of patients in a small series had a decreased DLCO. In this study the value of DLCO correlated with the duration of the disease, but the cause of the impaired diffusing capacity was not explored. Some of this group may have had a proliferative vasculopathy associated with MCTD, but other possibilities include fibrotic lung disease and emphysema. Patients with MCTD have been reported as having functional small airway obstruction, but there is no high-resolution CT (HRCT) or pathologic confirmation of this finding. However, large airway disease has been described in a single HRCT study that reported bronchiectasis in a few individuals.
Manifestations of the Disease
Radiography
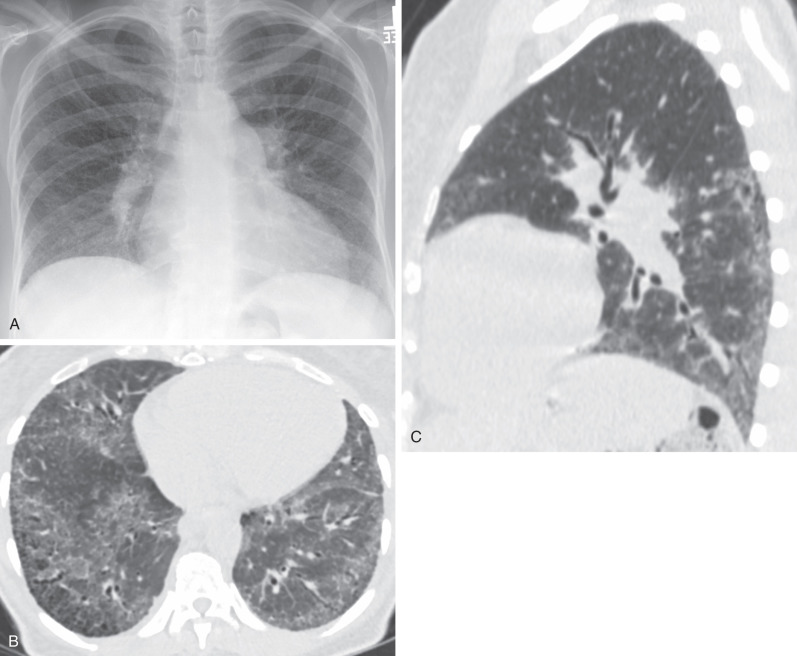
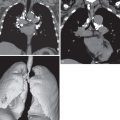
The true burden of lung disease in MCTD is unclear. On chest radiography interstitial lung disease has been reported to affect between 21% and 85% of patients. The typical radiographic abnormality is a reticular or reticulonodular pattern in the lower zones ( Fig. 44.1 ). Pleural effusions or thickening is seen on less than 10% of chest radiographs. Because these patients are often immunosuppressed, it is important to rule out infection in the face of nonspecific pulmonary abnormalities. Chest radiographs are less sensitive than HRCT for the detection of interstitial lung disease, and an apparently normal radiograph does not rule out interstitial lung disease.
Computed Tomography
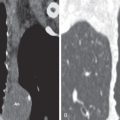
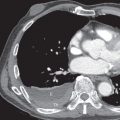
Nonspecific interstitial pneumonia (NSIP) is the most frequent pattern of interstitial lung disease in MCTD; UIP and lymphocytic interstitial pneumonia are less commonly observed patterns. One study of MCTD reviewed 41 HRCT scans with lung abnormalities in patients with MCTD. Most patients had an NSIP pattern of disease, with micronodules, ground-glass opacities, and intralobular lines (reticulation), predominantly in the lower zones (see Fig. 44.1 ). Nearly 50% of scans showed evidence of fibrosis, including intralobular lines, architectural distortion, and traction bronchiectasis. A small proportion of patients had airspace consolidation, honeycombing, cysts, and bronchiectasis resembling a UIP pattern of fibrosis ( Fig. 44.2 ). Therefore the HRCT manifestations of MCTD include findings seen in patients with systemic sclerosis, systemic lupus erythematosus, and polymyositis/dermatomyositis.

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


