(1)
Neurobiologie des processus adaptatifs, UMR7102, Université Pierre et Marie Curie, Paris, France
Abstract
Genetically encoded indicators are valuable tools to study intracellular signaling cascades in real time using fluorescent or bioluminescent imaging techniques. Imaging of Ca2+ indicators is widely used to record transient intracellular Ca2+ increases associated with bioelectrical activity. The natural bioluminescent Ca2+ sensor aequorin has been historically the first Ca2+ indicator used to address biological questions. Aequorin imaging offers several advantages over fluorescent reporters: it is virtually devoid of background signal; it does not require light excitation and interferes little with intracellular processes. Genetically encoded sensors such as aequorin are commonly used in dissociated cultured cells; however it becomes more challenging to express them in differentiated intact specimen such as brain tissue. Here we describe a method to express a GFP-aequorin (GA) fusion protein in pyramidal cells of neocortical acute slices using recombinant Sindbis virus. This technique allows expressing GA in several hundreds of neurons on the same slice and to perform the bioluminescence recording of Ca2+ transients in single neurons or multiple neurons simultaneously.
Key words
AequorinCalcium imagingNeocortexSindbisBioluminescence1 Introduction
1.1 Bioluminescent Genetically Encoded Ca2+ Indicator
Aequorin, isolated from jellyfish Aequorea species, is a bioluminescent complex that emits blue light upon Ca2+ binding [1]. Aequorin was first used as a Ca2+ indicator to evidence the role of intracellular Ca2+ in excitation–contraction coupling [2], neuronal signaling [3, 4], and meosis [5]. Since then, aequorin has been extensively used as a reporter of Ca2+ physiology in various cell types and subcellular compartments following intracellular injection or expression by gene transfer (reviewed in ref. 6).
Aequorin belongs to the family of photoproteins, i.e., it is a stable luciferase intermediate formed from the reaction of the protein apoaequorin (luciferase) and the prosthetic group coelenterazine (luciferin), and contains three EF-hand Ca2+-binding sites [7–9]. The formation of the aequorin complex is a slow process, whereas the bioluminescence reaction occurs as a rapid flash whose intensity increases with Ca2+ concentration and proceeds to completion in the continuous presence of Ca2+ [7, 10–12]. The rapid kinetics of Ca2+ binding to and unbinding from aequorin makes it a suitable indicator of rapid Ca2+ transients [10]. Aequorin Ca2+ sensitivity span concentrations from 0.1 to 1 mM [12, 13]. In addition, aequorin mutants and semisynthetic aequorins (i.e., incorporating synthetic coelenterazine analogue) endowed with different kinetics, Ca2+ sensitivity or spectral properties [12, 14–17] expand the range of bioluminescent Ca2+ sensors available to diverse applications [18–20].
A GFP-aequorin (GA) fusion protein has been described that allows fluorescence labeling of expressing cells and bioluminescence Ca2+ imaging of single cultured cells, of tissue slices and in whole vertebrate and invertebrate animals [21–25]. The fact that no illumination is required has allowed the use of GA to report neuronal activity in freely behaving zebrafishes [23]. No conspicuous morphological or functional alteration has been observed upon acute high-level expression of GA or in transgenic animals stably expressing GA [22–24]. Its bioluminescence in cellulo is superior to that of aequorin alone, presumably because the latter is rapidly degraded in the cytoplasm [26]. Bioluminescence resonance energy transfer between the photoprotein and GFP moieties of both fusion proteins shifts light emission to the green [27, 28] as observed between native fluorescent proteins and photoproteins in light emitting cells of Aequorea victoria [29]. Further shift in emission spectra has been observed when aequorin was fused to yellow or red fluorescent proteins enabling light detection though intact skin on living animals [30, 31].
Bioluminescence imaging with photoproteins is endowed with a high signal-to-noise ratio. Indeed, photoproteins exhibit negligible Ca2+-independent luminescence and their response intensity varies by several orders of magnitude depending on Ca2+ concentration [13, 32, 33]. GFP-photoproteins behave as low affinity, supralinear indicators of Ca2+ transients associated with action potentials in mammalian neurons [21]. A detection threshold of five action potentials has been reported in cortical neurons upon bioluminescence imaging in brain slices, corresponding to Ca2+ concentration transients locally reaching the micromolar range [21]. This detection threshold is comparable to that reported for the GCaMP genetically encoded fluorescent Ca2+ sensor [34]. The substrate coelenterazine is generally loaded once for reconstitution of active photoprotein prior to imaging experiments. The absence of coelenterazine during the course of the experiment eventually leads to exhaustion of the active photoprotein. However, due to the relatively small percentage of aequorin consumed, long duration continuous recordings of several hours have been reported [23].
1.2 Sindbis-Based Gene Transfer into Neurons
Sindbis virus belongs to the family of alphaviruses, in that its genome consist of a single-stranded, plus-strand RNA genome with a 5′ cap and a 3′ poly(A) tail. Upon entry into the cell, the viral RNA is directly translated by the cell host machinery and a virally encoded RNA-dependent RNA polymerase amplifies the genomic RNA, and generates transcripts that are under the control of a subgenomic promoter within the genomic RNA.
SIN virus has been successfully transformed into expression vectors, in which the transgene in the “vector RNA” is located downstream the subgenomic promoter and replaces the viral structural protein genes [35]. Shortly after cell transduction, the subgenomic RNA becomes the most abundant message in the transduced cells and promptly recruits most of the host’s translational machinery for its own use, resulting in high levels of the desired protein molecules in the cytoplasm few hours after transduction. Such expression SIN vectors are called pseudovirions as they do not encode for structural proteins such as capsid and have little or no plaque forming unit capability. For more details about the biology of Sindbis vectors see ref. [36].
Compared to other viral vectors, advantages of SIN vectors are available plasmids for cloning the transgene and synthesis of the recombinant vector RNA, large insert size (6.5 kb), easy and fast generation of recombinant viral particles, high viral titers obtained, rapid onset, and high-level transgene expression (within hours) [37]. When applied to neuronal tissue, it possesses the additional advantage of efficiently and preferentially transducing neurons rather than nonneuronal cells [36]. In recent years, Sindbis viruses have been used to express various membrane receptors, anchoring molecules and kinases in cultured neurons and cultured slices, and also in vivo [36]. Furthermore, several genes can be expressed separately using a multiple subgenomic promoter [38].
Aside from their advantages, wild-type SIN pseudovirion vectors [35] retain the disadvantage of inhibiting host cell protein synthesis, which eventually causes cell death. Therefore, SIN expression systems are often used for transient expression experiments. While neurotoxic effects appear relatively fast (within 1–2 days), some mutation in the nonstructural protein 2 or the use of optimized helper RNA have been found to delay the onset of cellular toxicity by several days [39, 40]. In addition of SIN pseudovirions, other expression vectors based on the semliki forest virus (SFV), another alphavirus, have been successfully assembled [36] which allow targeting either a specific neuronal cell types or nonneuronal cells [36]. This can be achieved for example by engineering coat protein [41] or using microRNA [42].
In this chapter we describe a protocol to prepare acute slice for SIN viral infection and to perform subsequent imaging of the GA light response upon neuronal activity evoked by electrical stimulation. It must be noted that we routinely use SIN pseudovirions generated using the vector plasmid pSinRep5 [35] to express several types of genetically encoded indicators such as GA into neocortical pyramidal cell or thalamic neurons of acute slices [21, 43–45].
2 Materials
Experimental animals: rats (Wistar) or mice (C57/Bl6). All animal experiments are to be performed in accordance with the guidelines on the use of animals by the relevant authorities.
CAUTION: The low level of pathogenicity of SIN virus in humans has allowed it to be classified as a Biosafety Level-2 (BL-2) agent by the NIH Recombinant DNA Advisory Committee. Recommended precautions include standard microbiological practices, laboratory coats, inactivation of all infectious waste, limited access to working areas, protective gloves, posted biohazard signs, and class I or II biological safety cabinets used for mechanical and manipulative procedures that cause splashes or aerosol. All personnel working with the SIN Expression System should be properly trained to work with BL-2 level organisms. SIN virus can be inactivated by organic solvents, bleach, or autoclaving. SIN vectors expressing a highly toxic protein should be treated as a special risk.
2.1 Preparation of Acute Slices
1.
10× ACSF (in mM): 1,260 NaCl, 25 KCl, 12.5 NaH2PO4, 260 NaHCO3. Weigh 146.1 g NaCl, 3.73 g KCl, 3 g anhydrous NaH2PO4, 43.7 g NaHCO3, and add to a 2 l-cylinder containing 1 l distilled water and make it up to 2 l. Mix well and store at 4 °C. This solution can be kept for 2 weeks.
2.
1× ACSF (in mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 20 d-glucose, and 5 Na pyruvate. Add in this order, 750 ml volvic water, d-glucose, 100 ml 10× ACSF, 2 ml CaCl2 1 M, 1 ml MgCl2 1 M and 0.55 g Na pyruvate. Adjust to 1 l and filter this solution on 0.22 μm membrane. This solution is prepared every day and saturated with carbogen (95 % O2, 5 % CO2) for 20 min before use.
3.
The cutting solution consists in ACSF supplemented with 1 mM kynurenic acid. It is also bubbled with carbogen. To help dissolving kynurenic acid, sonicate for 5–10 min the ACSF solution containing kynurenic acid using an ultrasound bath.
4.
Dissection tools for removing the brain. This includes Scissors for initial dissection, long thin scissors for removing skin, short scissors for cutting skull, short blunt forceps for removing skull, rounded spatula for removing brain, scalpel and blade for dividing brain, rounded spatula for transferring brain halves.
5.
Small parts: Adhesive cyanoacrylate (e.g., Loctite 406), Stainless steel blade or razor blade, plastic Petri dishes (diameter 35 mm), Pasteur pipette for transferring slices.
6.
Anesthetics ketamine (100 mg/kg body weight) and xylasine (10 mg/kg body weight). In a 5 ml tube, add 125 μl xylasine (e.g., Rampun 0.2 %), 250 μl ketamine (e.g., Imalgene 1000), and 2.125 ml NaCl 0.9 %. Store this solution at 4 °C.
2.2 Slice Culture
1.
In a 500 ml cylinder, add 250 ml Minimal essential medium (MEM), 250 ml 1× Hanks balanced salt solution (HBSS), 5 ml Penicillin–streptomycin (P: 10,000 U/ml, S: 10,000 mg/ml), 2.75 g d-glucose (6.5 g/l final) then filter-sterilized before aliquoting in 50 ml tube (see Note 1).
2.
Millicell culture plate inserts (Millipore); these inserts have low walls, which help to the handling of the slices and allow putting the insert in a 35 mm Petri dish.
3.
Recombinant Sindbis pseudovirus. A detailed protocol for the production of Sindbis-based vectors can be found elsewhere [46]. We routinely use SIN vectors assembled using pSinRep5 vector plasmids and the DH26S helper plasmid [35]. Briefly, the coding sequence of the gene of interest is inserted in pSinRep5 downstream of the subgenomic promoter. The resulting plasmids and pDH26S are then linearized and in vitro transcribed into capped RNA using SP6 polymerase. Both RNAs are electroporated into BHK-21 cells and supernatant containing SIN pseudovirion is collected 24 h later.
4.
Native coelenterazine free-base, stock solution at 1.25 mM in ethanol. Stored at −80 °C. Caution: manipulate coelenterazine in the dark.
2.3 Equipment
1.
Laminar flow cabinet and a 5 % CO2 incubator maintained at 35 °C.
2.
High-quality vibrating slicer (VT1000S or VT1200; Leica Microsystems).
3.
Incubation chamber: a submerged chamber optimized to ensure sufficient oxygenation of the tissue during the recovery period. Usually, it consists of a small plastic cylinder ending in a nylon net.
4.
Grid of nylon threads glued to a U-shaped platinum frame to maintain the slice in the bottom of the recording chamber. The platinum U-frame should be centered to avoid any mechanical interaction between its walls and the recording pipettes.
5.
An imaging setup that can contain an upright microscope with water immersion objective, a recording chamber and a perfusion system. Example of such equipment can be found elsewhere [47, 48]. We are using an intensified CCD video camera (ICCD225; 768 × 576 pixels; Photek, St Leonards on Sea, UK) mounted on the C-mount port of an upright BX51WI microscope (Olympus) and controlled by the data acquisition software IFS32 (Photek). Light acquisition is performed through water immersion objectives 10× (N.A. = 0.3) and 60× (N.A. = 0.9). The imaging setup (see Fig. 1) is housed in a dark box to avoid light noise. Slices are set in a recording chamber that is continuously perfused with standard artificial cerebro-spinal fluid (ACSF). GFP fluorescence is visualized using a mercury lamp with a standard GFP filter set (e.g., from Chroma technology, cat no 41018, or Semrock, cat no GFP-30LP-B-000). Filters are removed from light path during bioluminescence recordings.
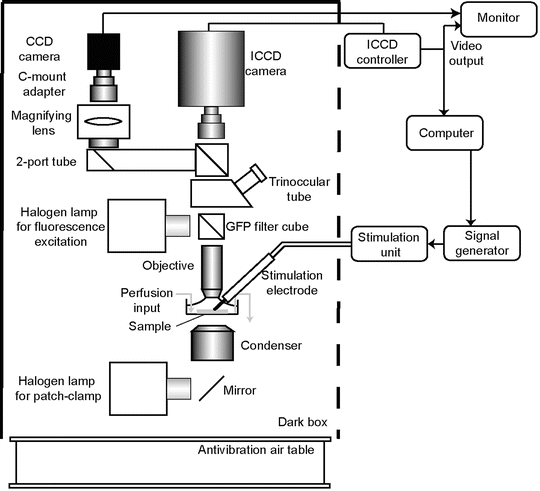
Fig. 1
Schematic representation of a setup for bioluminescence recordings. Prior to imaging, samples are visualized by GFP epifluorescence using GFP filter set. Filters reside in a filter wheel (not shown), thus they can be removed from the light path during bioluminescence acquisition. Fluorescence image can be acquired either with the low gain mode of the ICCD camera or using a regular CCD camera connected to a 2-tube port
6.
For electrical stimulation, a concentric bipolar electrode (SNEX-100, Rhode biomedical, tip end 100 μm wide) is set up on a manually driven micromanipulator (Mini 25 3Axes, Luigs and Neuman, Germany). The electrode is connected to a constant current stimulation unit like the DS2A from Digitimer (UK). Pulse amplitude and duration are set on the stimulation unit. The stimulus wave form is provided by a TTL signal either by the software pClamp through a digidata A/D converter or with a signal generator (Accupulse, World Precision Instrument).
3 Methods
3.1 Brain Dissection and Acute Slice/Slice Culture Preparation
1.
Prepare the slicing setup by putting 250 ml of cutting solution in the freezer until ice appears (see Note 2). Transfer this solution to a polystyrene box containing ice and saturate the liquid/ice mixture with carbogen for 20 min. During that period, install dissection instruments on the bench, set up the razor blade on the blade holder of the slicing microtome and put some ice around the slicing chamber in which the slice will be cut. Some water can be added to the ice to facilitate the cooling of the tray and to keep it cold along the slicing procedure. Prepare an inverted suction pipette to water the brain with a Pasteur pipette broken at the narrow tip and mounted on a rubber bulb.
2.
 Blood-Based Assay with Secreted Gaussia Luciferase to Monitor Tumor Metastasis
Blood-Based Assay with Secreted Gaussia Luciferase to Monitor Tumor Metastasis
 Bioluminescent Imaging of Bacteria During Mouse Infection
Bioluminescent Imaging of Bacteria During Mouse Infection
 Noninvasive In Vivo Monitoring of Extracellular Vesicles
Noninvasive In Vivo Monitoring of Extracellular Vesicles
 Bioluminescence Imaging of Fungal Biofilm Development in Live Animals
Bioluminescence Imaging of Fungal Biofilm Development in Live Animals
 Monitoring of Transcriptional Dynamics of HIF and NFκB Activities
Monitoring of Transcriptional Dynamics of HIF and NFκB Activities




Related posts:
Blood-Based Assay with Secreted Gaussia Luciferase to Monitor Tumor Metastasis
Bioluminescent Imaging of Bacteria During Mouse Infection
Noninvasive In Vivo Monitoring of Extracellular Vesicles
Bioluminescence Imaging of Fungal Biofilm Development in Live Animals
Monitoring of Transcriptional Dynamics of HIF and NFκB Activities
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
