Etiology, Prevalence, and Epidemiology
Nonspecific interstitial pneumonia (NSIP) is a chronic interstitial lung disease characterized by homogeneous expansion of the alveolar walls by inflammation or fibrosis or both. NSIP is the second most common chronic interstitial pneumonia, after usual interstitial pneumonia (UIP), accounting for 14% to 35% of cases. NSIP may be idiopathic but more commonly occurs as a manifestation of connective tissue disease, hypersensitivity pneumonitis, drug-induced lung disease, and chronic interstitial lung disease complicating diffuse alveolar damage.
The prognosis of NSIP is influenced by its predominant histologic component. Patients with a predominantly or exclusively inflammatory component (i.e., cellular NSIP) have an excellent prognosis with few reported deaths, whereas the reported median survival of patients with fibrotic NSIP ranges from approximately 6 to 14 years. The prognosis of NSIP is considerably better than the prognosis of idiopathic pulmonary fibrosis (IPF), the clinical condition most commonly associated with UIP.
Clinical Presentation
The median age of onset of symptoms in NSIP is 40 to 50 years, which is more than 10 years younger than patients with IPF. NSIP may occur in childhood and in the elderly, however, having been reported in patients 9 to 78 years old. The symptoms, similar to those of IPF, consist of progressive dyspnea and dry cough with a duration ranging from 6 months to 3 years before diagnosis. Finger clubbing occurs in 10% to 35% of patients, which is less common than in patients with IPF. Auscultation reveals basal or widespread crackles.
Pathophysiology
Pathology
The pathogenesis of NSIP is unknown. Because it is commonly seen in patients with connective tissue disease, especially systemic sclerosis, many investigators believe that NSIP is an autoimmune disease. It also may be a subclinical form of other nonidiopathic interstitial lung diseases, such as hypersensitivity pneumonitis.
NSIP is characterized histologically by homogeneous expansion of the alveolar walls by inflammation or fibrosis or both ( Fig. 28.1 ). The temporal and geographic homogeneity of NSIP, with the findings seeming to represent the same stage in the evolution of the disease, is distinct from the heterogeneity seen in UIP. The histologic findings may range from inflammation (i.e., cellular NSIP) to predominant fibrosis (i.e., fibrotic NSIP). In cellular NSIP the alveolar septa are thickened by infiltrates of lymphocytes and plasma cells, whereas in fibrotic NSIP the thickening is mainly due to collagen accumulation. The extent of interstitial fibrosis varies. The fibrosis may involve the alveolar septa, peribronchiolar interstitium, interlobular septa, and visceral pleura. Areas of organizing pneumonia may be seen but are usually focal and mild. Fibroblastic foci (i.e., aggregates of proliferating fibroblasts and myofibroblasts that give UIP the appearance of temporal heterogeneity) are absent or inconspicuous.

The histologic differential diagnosis of NSIP includes hypersensitivity pneumonitis, lymphoid interstitial pneumonia, organizing pneumonia, and UIP. In contrast to NSIP, the interstitial pneumonia in hypersensitivity pneumonitis has a bronchiolocentric distribution. Other helpful findings are the presence of chronic bronchiolitis and poorly formed granulomas, seen in 50% to 60% of patients with hypersensitivity pneumonitis. Lymphoid interstitial pneumonia is similar to NSIP, except that it is typically associated with lymphoid hyperplasia in the form of peribronchiolar lymphoid aggregates. Poorly sampled cases of cryptogenic organizing pneumonia may be misdiagnosed on biopsy as being NSIP. A more difficult differential diagnosis is between NSIP and UIP because focal areas indistinguishable from NSIP commonly occur in what are otherwise typical cases of UIP. Histologically, if one section has features diagnostic of UIP, and another section shows NSIP, the final diagnosis is UIP. Nonetheless, there is considerable interobserver variability even among expert histopathologists in the diagnosis of NSIP and in the distinction of fibrotic NSIP from UIP.
Lung Function
NSIP results in restrictive lung function (decreased total lung capacity and vital capacity) and impaired gas exchange as assessed by the carbon monoxide diffusing capacity. The functional impairment varies but tends to be milder than in patients with IPF.
Manifestations of the Disease
Radiography
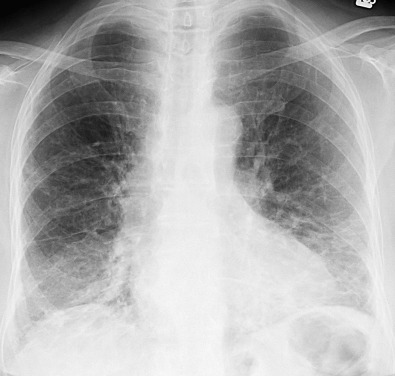
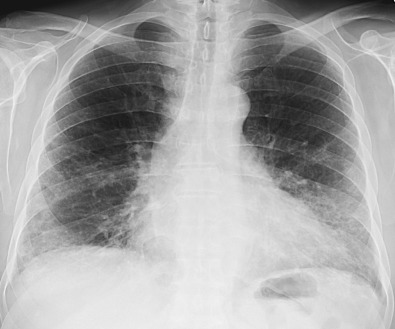
The most common radiographic abnormalities are bilateral patchy or confluent hazy areas of increased opacity (ground-glass opacities) involving mainly the middle and lower lung zones ( Fig. 28.2 ). Other manifestations include a reticular pattern or a combination of reticular opacities, ground-glass opacities, and consolidation ( Fig. 28.3 ). In 15% of patients with NSIP and abnormal high-resolution computed tomography (CT) findings, the chest radiograph is normal.
Computed Tomography
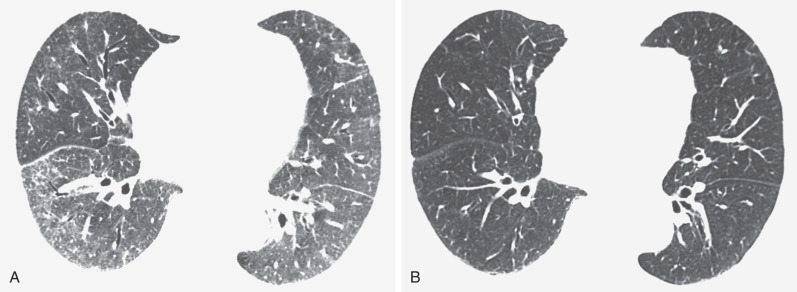
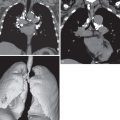
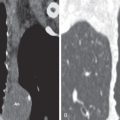
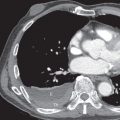
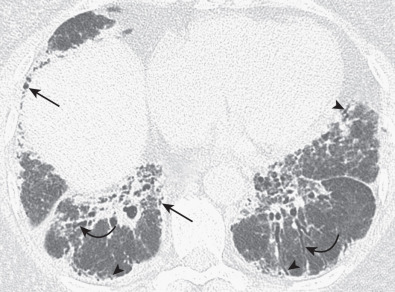
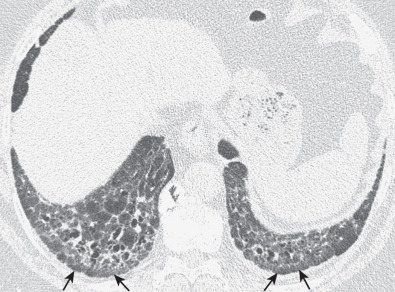
The most common high-resolution CT manifestation of NSIP consists of bilateral symmetric ground-glass opacities ( Figs. 28.4 and 28.5 ). Most patients have a fine reticular pattern and traction bronchiectasis superimposed on the ground-glass opacities ( Fig. 28.6 ). The prevalence of reticular opacities reported in various studies ranges from 50% to 100% of cases. Honeycombing is seen in 10% to 30% of patients and, when present, tends to be mild, involving less than 10% of the parenchyma ( Fig. 28.7 ). Areas of consolidation and centrilobular nodules have been reported in a small percentage of patients in some studies but were not seen in one other large study. The areas of consolidation may correspond to focal organizing pneumonia and are seen most commonly in patients with collagen vascular disease. The abnormalities in NSIP may be diffuse but in 60% to 90% of cases involve mainly the lower lung zones and in 50% to 70% of patients involve predominantly the peripheral lung regions ( Fig. 28.8 ). A combination of predominant peripheral and peribronchovascular distribution is present in approximately 10% of patients. Although the distribution of NSIP is frequently peripheral, it has been shown that in approximately 50% of patients with fibrotic NSIP the fibrosis tends to spare the lung immediately adjacent to the pleura at two or more levels in the dorsal regions of the lower lobes ( Figs. 28.9 and 28.10 ). This “relative subpleural sparing” is highly specific for fibrotic NSIP; conversely, UIP typically is most severe in the subpleural regions.
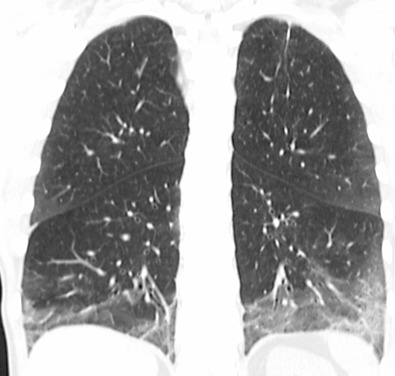
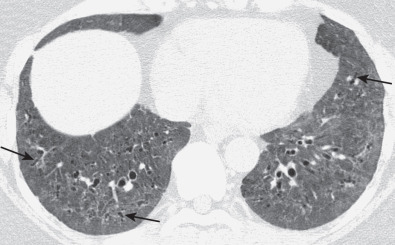
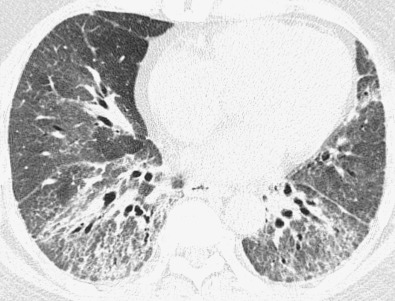
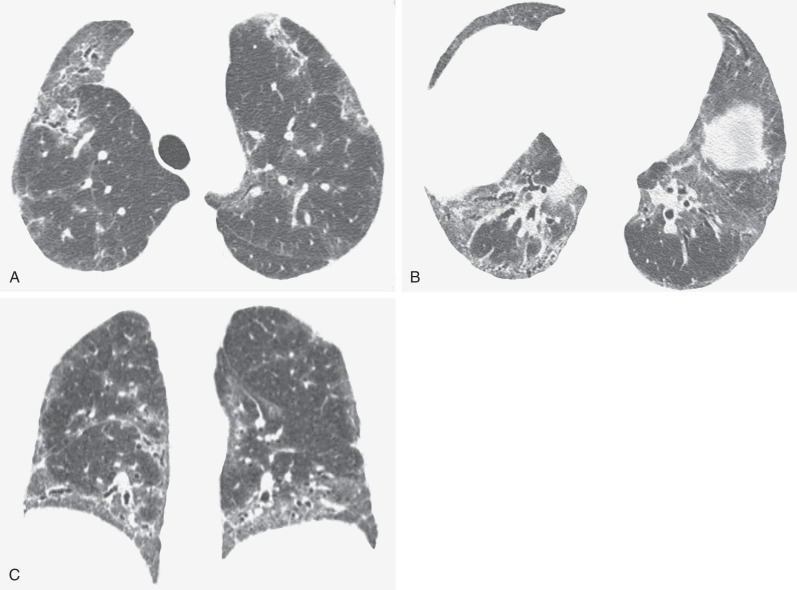
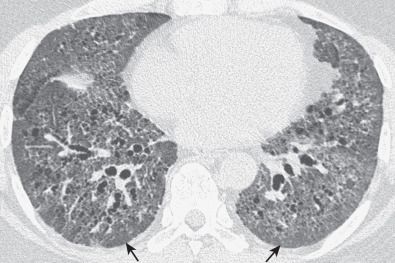
Patients with NSIP and only ground-glass opacities on high-resolution CT typically have cellular NSIP (see Fig. 28.4 ). Patients with ground-glass opacities, reticulation, and traction bronchiectasis may have cellular (inflammatory) or fibrotic NSIP (see Figs. 28.5 to 28.10 ). In one study the authors compared high-resolution CT findings of the subtypes of NSIP in 55 patients. There was no appreciable difference in the extent of ground-glass opacities (average extent, 30% of the parenchyma), consolidation (average extent, 10%), small nodules (average extent, 9%), or interlobular septal thickening (average extent, 5%) between cellular and fibrotic NSIP. Patients with fibrotic NSIP had greater extent of intralobular lines (reticulation) (12% vs. 8%) and traction bronchiectasis (15% vs. 5%) than patients with cellular NSIP. Mild honeycombing was seen on high-resolution CT in 13 of 33 (39%) patients with fibrotic NSIP. In a second study the investigators compared the high-resolution CT findings in 6 patients with predominantly cellular NSIP, 15 patients with equivalent extent of inflammation and fibrosis, and 15 patients with predominantly fibrosis on lung biopsy. The extent of ground-glass opacities in the three groups was similar, with an average extent ranging from 25% to 35% of the lung parenchyma. All three groups had an average extent of consolidation of 10%. The extent of intralobular linear opacities (reticular pattern) ranged from an average of 13% of the parenchyma in patients with predominant inflammation to an average of 23% in patients with predominant fibrosis. Honeycombing was not seen in any patient with predominant inflammation or equivalent inflammation and fibrosis but was seen in patients with predominant fibrosis (average extent, 4%).
Serial CT scans in patients with NSIP have shown that patients with predominant ground-glass opacities on the initial CT scan are more likely to improve with treatment and have a better long-term prognosis than patients with predominant fibrosis ( Figs. 28.11 to 28.13 ). In one study of 13 patients with biopsy-proven NSIP, the initial CT scans showed ground-glass opacities and, to a lesser extent, reticulation. On follow-up high-resolution CT there was a significant reduction in the extent of ground-glass opacities and an improvement in forced vital capacity that correlated with the extent of reduction in ground-glass opacities on CT. Similar results were described in a study of 14 patients with NSIP associated with polymyositis or dermatomyositis. In a third study serial CT scans were performed in 38 patients with histologically proven NSIP, including 4 with cellular NSIP, 13 with mixed cellular and fibrotic NSIP, and 21 with fibrotic NSIP. The predominant initial CT pattern was inflammatory (ground-glass opacities and consolidation) in 6 (16%) patients and fibrotic (reticulation and honeycombing) in 32 (84%) patients. The predominant pattern on initial high-resolution CT was significantly associated with change in extent of parenchymal abnormality on follow-up CT. At a mean follow-up of approximately 1 year, all of the patients with an inflammatory-predominant pattern on the initial CT scan improved, whereas of the 32 patients with a fibrosis-predominant pattern, 7 (22%) improved, 6 (19%) deteriorated, and 19 (59%) remained stable. There was no significant association between the histologic findings and the likelihood of improvement on follow-up CT.