OPTIC NERVE AND SHEATH: BENIGN AND MALIGNANT TUMORS
KEY POINTS
- Computed tomography and magnetic resonance imaging in combination are the primary tools, far more so than biopsy, for establishing the diagnosis of optic pathway glioma and optic nerve sheath meningioma.
- Computed tomography and magnetic resonance imaging are in the critical pathway for evaluating the cause of a visual loss that might be due to compression or other involvement of the optic nerve and sheath.
- Computed tomography and magnetic resonance imaging are extremely useful in narrowing the differential diagnosis of disease that may affect the optic nerve and sheath.
INTRODUCTION AND GENERAL DIAGNOSTIC CONSIDERATIONS
Lesions of either the optic nerve or optic sheath may spread along the longitudinal axis of the nerve, be locally exophytic, or show a combination of these features. The exophytic form would be atypical for nerve lesions and more likely associated with sheath tumors. A vector along the nerve suggests an intra-axial origin, such as an optic pathway glioma (OPG); an infiltrative neoplasm such as lymphoma; or an inflammatory lesion such as optic neuritis from multiple sclerosis, syphilis, or Wegener granulomatosis (Chapter 55). The inflammatory and non–nerve-origin neoplasm may also show a pia-arachnoid spread pattern on the surface of the nerve and separate from the inner aspect of the sheath.
Differentiation between extra-axial and intra-axial disease requires demonstration of a margin between the mass and optic nerve. If a margin exists, then the lesion very likely originates in or outside the sheath. If no margin exists and the mass is well circumscribed, then the lesion probably arises within the nerve primarily; however, more infiltrative processes possibly involving the entire nerve/sheath complex may blur this distinction. Accurate diagnosis typically requires a combination of coronal and axial imaging as well as consideration of other imaging findings than those directly affecting the nerve and sheath. Accurate assessment of the findings also requires careful consideration of clinical factors such as age, visual symptoms, and the presence of a known systemic disease. Sometimes, these conditions must be distinguished from an optic sheath “meningocele.”1
Dilatation of the optic subarachnoid space or optic hydrops can be a variation of normal or seen as part of communicating hydrocephalus, the latter perhaps a clue to disseminated intracranial pathology. Cellular infiltration of the subarachnoid space can result in such optic sheath hydrops due to the spread of tumors or other disease such as sarcoidosis and tuberculosis intracranially. Carcinomatosis of the intracranial subarachnoid spaces may also seed the optic nerve via this pathway, resulting in dilatation of the sheath with little clear evidence as to the etiology.
ANATOMIC AND DEVELOPMENTAL CONSIDERATIONS
Applied Anatomy
The anatomy of the optic nerve and sheath in relation to the eye and orbit as well as more posterior visual pathways are discussed in detail in Chapter 44.
In summary, the optic nerve brings with it layers of pia mater, arachnoid, and dura, and by necessity, subarachnoid and subdural spaces, as it exits the cranial vault via the optic canal (Figs. 44.15, 44.19, 44.22, and 44.23) to eventually join the eye. Any of its segments from the eye to the brain may be involved in these neoplastic processes, some of them being dural or leptomeningeal in origin rather than primarily arising from the optic nerve.
IMAGING APPROACH
Techniques and Relevant Aspects
The eye and optic nerve and sheath are studied with ultrasound (US), computed tomography (CT), and magnetic resonance (MR) techniques described in detail in Chapters 44 and 45. The tumors involving the optic nerve and sheath may be seen coincidentally on images of the brain and face and, therefore, on images with much lower resolving power than when those studies might be focused on the eye and optic nerve or at least the orbit.
Dedicated studies of the eye/orbit must be done with the highest possible resolution given other constraints on all of the anatomy that needs to be evaluated. Often, a separate protocol for the eye, optic nerve, and orbit is required if both the brain and eye/orbit must be studied definitively with CT and/or magnetic resonance imaging (MRI).
Definitive imaging in this region requires careful technique because of the air–bone interfaces surrounding the optic canal and the small size of the optic nerve/sheath complex. On CT, the bone density may obscure contrast enhancement. On MR, air within the sinuses creates susceptibility artifacts, and fatty marrow within the clinoid process can obscure contrast enhancement. Therefore, fat-suppressed and non–fat-suppressed T1-weighted images with and without gadolinium, in multiple planes, are focused on the orbital apex, optic canal, and cistern in the evaluation of this region.
Specific CT and MRI protocols by indications are detailed in Appendixes A and B, respectively. Almost all CT and MRI studies are done with contrast.
CT gantry angulation has been in the past changed from the usual zero degrees to the infraorbital meatal line to −10 degrees for emphasis on the course of the optic nerve to and then through the optic canal. This is typically no longer done with workstations now capable of rapid off-axis viewing of volume data sets. Acquisition slice thickness should be ≤0.5 to 1.0 mm. Reformations will be of excellent quality with such data acquisition.
Pros and Cons
Tumors of the optic nerve and sheath are studied primarily with MRI and usually with CT done as an adjunct. MRI is far more definitive than CT in its rendering of the optic nerve as separate from the optic sheath. MRI is also far more sensitive than CT to meningeal pathology. It detects intracranial involvement with these neoplastic processes and their intracranial complications much better than CT. Dilatation of the nerve/sheath complex (optic sheath hydrops) on CT can mimic a diffusely infiltrative process; whereas on MR, the cerebrospinal fluid (CSF), nerve, and pathology are much more likely to be distinguished.1,2
CT is used adjunctively or in patients too sick or for some other reason unable to complete a very high quality MRI examination. It is also used to identify possible bone findings that might contribute to medical decision making, such as help in the differential diagnosis or a plan for gross total resection. CT is a reasonable secondary screening tool for identifying intracranial extension of disease and intracranial complications. Computed tomographic angiography is an adequate screening tool to confirm vascular encasement and cavernous sinus involvement when MR cannot be done.
US and radionuclide studies are not generally used to study these lesions.
SPECIFIC DISEASE/CONDITION
Optic Pathway Glioma
Etiology
OPG is a brain tumor involving the optic nerve(s), the optic chiasm, and/or optic tract(s). Lesions may progress slowly or not at all, and some can actually regress with time (Figs. 29.21, 29.22, and 56.1 and Chapter 29). They frequently present as solid masses in infants younger than 2 years of age. Bilateral hamartomatous optic gliomas have a very strong association with neurofibromatosis type 1 (NF-1) and hybrid phacomatoses (Figs. 29.21, 29.22, and 56.1 and Chapter 29). Unilateral disease is more often sporadic than associated with NF-1, but the risk of NF-1 is high even with unilateral OPG in a child.
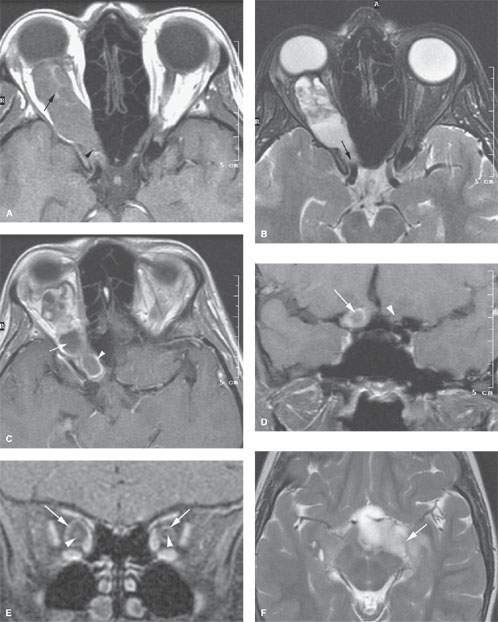
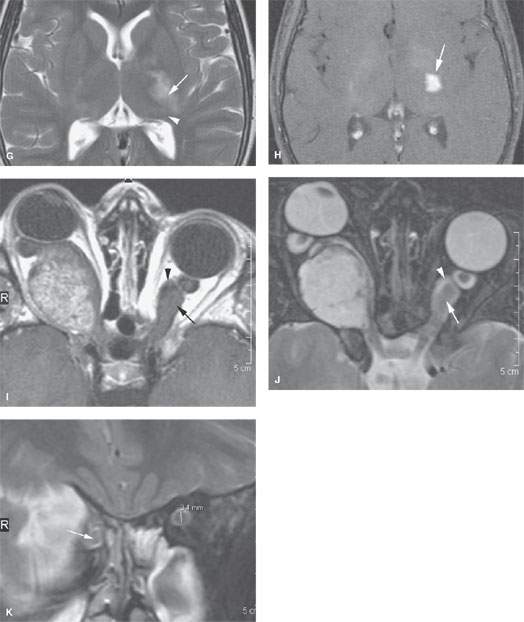
FIGURE 56.1. Magnetic resonance imaging studies of four patients with optic pathway gliomas (OPGs). Additional images of OPGs are shown in Figs. 29.21 and 29.22. A–D: Patient 1. In (A), a contrast-enhanced T1-weighted (T1W) image of a bulky OPG on the right with minimal enhancement presents centrally (arrow). In (B), the T2-weighted (T2W) image shows the inhomogeneity of signal within the mass (arrow) and that it might extend to the optic chiasm as in (A). In (C), the contrast-enhanced T1W image shows a relatively unusual morphology with broad zones of nonenhancing, cystic change (arrow) and peripheral enhancement (arrowhead). In (D), the T1W coronal image shows that the lesion has extended to the cranial portion of the optic nerve (arrow) compared to the normal nerve on the opposite side (arrowhead). The optic chiasm was not involved. E–H: Patient 2. In (E), the T1W fat-suppressed image shows a relatively unusual pattern of enhancement in these bilateral OPGs, that being a central (arrowheads) and peripheral zone (arrows) of enhancement in a markedly enlarged nerve on the right and one of relatively normal size on the left. In (F), T2W images show that while the involvement of the nerve on the left side seemed lesser, there was more involvement at the point of entry of the optic tract into the brain (arrow). In (G), the T2W image shows continued extension along the posterior optic pathways (arrow) toward the lateral geniculate body (arrowhead). In (H), extension along the posterior visual pathways shows enhancement (arrow) consistent with a more aggressive pattern of disease and consistent with pilocytic astrocytoma than hamartomatous changes along the visual pathways. I, J: Patient 3. In (I), the contrast-enhanced T1W image shows what appears to be a relatively homogeneously enhancing, possibly even exophytic, lesion on the right side, but this is an OPG. This is an obvious diagnosis once the dilatation of the left optic sheath (arrowhead) and enlargement of the optic nerve (arrow) is identified. In (J), the T2W image confirms the findings in (I). K: Patient 4. A patient with obvious stigmata of neurofibromatosis type 1 and an OPG on the right (arrow). Clinically, it was uncertain whether there were bilateral optic nerve abnormalities. The left optic nerve measured 3.4 mm, well beyond range of normal variation, indicating that bilateral disease was present.
Prevalence and Epidemiology
OPG occurs with a bimodal distribution in the young and in older adults. Up to 40% of children with NF-1 may develop an OPG, and a similar percentage of children presenting with OPG will subsequently be diagnosed with NF-1. It has no particular gender predilection.
Clinical Presentation
Patients will usually present with painless proptosis. Visual loss is a possible late symptom. Often, an afferent pupillary defect will be present. In large lesions at the chiasm, pituitary or hypothalamic dysfunction is possible. Optic gliomas characteristically display an intra-axial enlargement of the optic nerve by the time of presentation.
Pathophysiology and Patterns of Disease
OPG has a wide range of biologic activity and histopathologic appearances, ranging from hamartomatous lesions in neonates to a pilocytic astrocytoma in a child to a glioblastoma in an older adult (Figs. 29.21, 29.22 and 56.1 and Chapter 29). Bilateral hamartomatous optic gliomas have a strong association with NF-1.
These lesions may progress slowly or not at all, and some can actually regress with time. The optic canal will be enlarged when that segment of the nerve is involved (Figs. 29.21, 29.22, and 56.1 and Chapter 29). They are invariably solid masses found in infants younger than 2 years of age (Fig. 56.1). In children, an OPG is usually a slowly progressive pilocytic astrocytoma that will frequently involve all segments of the optic nerves and may grow across the midline and along the optic tracts to involve the brain (Figs. 29.21, 29.22, and 56.1 and Chapter 29).
Manifestations and Findings
Computed Tomography
The CT appearance of nerve sheath tumors and optic gliomas is discussed in detail in Chapter 29.
Optic gliomas characteristically display an intra-axial enlargement of the optic nerve by the time of presentation (Figs. 29.21, 29.22, and 56.1). CT changes may be subtle unless a difference in nerve/sheath complex size of about 2 to 3 mm is observed. In children, there is often diffuse tubular enlargement of the optic nerve with a kinked or bent appearance. More subtle changes may be due to optic sheath hydrops rather than the tumor itself. CT density alteration relative to the normal nerve/sheath complex seldom occurs, and unlike suprasellar optic gliomas, tumors in the nerves often have no abnormal enhancement, especially in children. In adults where high-grade tumors often exhibit detectable enhancement, more subtle lesions may be detectable with CT. Cystic changes may be detectable. Calcification is rare.
Magnetic Resonance
The MRI appearance of nerve sheath tumors and optic gliomas is discussed in detail in Chapter 29.
A small OPG is better detected by MR than CT imaging (Figs. 29.21, 29.22, and 56.1). On noncontrast T1-weighted images, the lesions tend to be isointense to the normal nerve and gray matter. On T2-weighted images, the tumors tend to be slightly to markedly hyperintense to white matter depending on their degree of differentiation. The more hamartomatous the lesion, the more likely it is to remain isointense to the brain on all pulse sequences. A pilocytic astrocytoma is more likely to show some cystic change and enhancement (Figs. 29.21, 29.22, and 56.1). Patterns of enhancement vary from homogeneous to patchy and do not necessarily indicate a poor prognosis.
An aim of imaging is to demonstrate the proximity of tumor to the optic chiasma; thus, very high resolution multiplanar images at the nerve–chiasm junction are a critical part of the study technique. Bilateral OPG without chiasm involvement is unusual1 (Fig. 56.1I,J).
Differential Diagnosis
From Supportive Diagnostic Techniques
The diagnosis is essentially made by imaging, and it is not often in doubt following a properly focused MRI study. The lesions are not biopsied. A strictly chiasmatic and retrochiasmatic lesion may be difficult to tell from a hypothalamic glioma.
Medical Decision Making and Treatment Options
Medical
The lesions are frequently just observed especially when they are of the hamartomatous variety. Chemotherapy may be used to help slow or stop their progression, and it is especially considered if the optic chiasm is threatened.
Surgical
Surgery is usually not a main part of the treatment plan. It may be used in a nonseeing eye to improve cosmesis. It is generally not used for chiasmatic and retrochiasmatic tumors. The diagnosis of OPG only rarely requires a biopsy for confirmation of the diagnosis.
Radiotherapy
Radiotherapy may be used to slow or stop tumor growth that is causing visual impairment to progress.
Surveillance Options
MRI is the standard mode of follow-up, whether to evaluate the effects of therapy or to monitor the response to therapy.
DISEASE ACUITY AND REPORTING RESPONSIBILITY
If an OPG is diagnosed, reporting is routine. If instead a compressive lesion of the optic nerve or other cause for visual loss is discovered, prompt communication is the best practice since prompt treatment may preserve vision that might otherwise be lost.
The report should clearly establish the extent of tumor within the optic nerve. It must establish its extent relative to the optic canal and optic chiasm, and especially spread approaching or across the midline that might threaten the contralateral optic nerve should be noted.
Related posts:
 Preseptal Compartment: Acute and Chronic Infections and Noninfectious Inflammatory Conditions
Preseptal Compartment: Acute and Chronic Infections and Noninfectious Inflammatory Conditions
 Intraconal Orbit: Tumors
Intraconal Orbit: Tumors
 Hypopharynx: Developmental Abnormalities
Hypopharynx: Developmental Abnormalities
 Tumors of the Submandibular Gland and Space and Tumorlike Conditions
Tumors of the Submandibular Gland and Space and Tumorlike Conditions
 Masticator Space, Buccal Space, and Infratemporal Fossa Infections and Other Inflammatory Conditions
Masticator Space, Buccal Space, and Infratemporal Fossa Infections and Other Inflammatory Conditions
 Oral Cavity and Floor of the Mouth: Malignant Tumors
Oral Cavity and Floor of the Mouth: Malignant Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


