Fig. 7.1
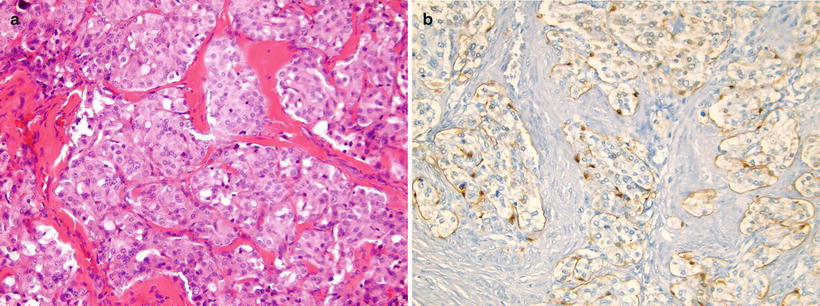
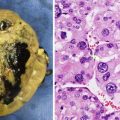
(a, b) Nasopharyngeal carcinoma. (a) Contrast-enhanced CT in a teenage boy demonstrating an asymmetric enhancing right nasopharyngeal with extension into the retropharyngeal soft tissues. A right maxillary retention cyst or mucosal polyp is incidentally noted. (b) Axial T1W postcontrast MRI image in the same patient shows the enhancing nasopharyngeal mass as well as delineates better the involvement of adjacent structures. Encasement of the carotid sheath vessels can be appreciated (arrow). (c) Contrast-enhanced CT image in a 12-year-old girl with an asymmetric enhancing nasopharyngeal mass similar in appearance to a nasopharyngeal carcinoma but confirmed by biopsy to be lymphoma. (d) Nasopharyngeal carcinoma, nonkeratinizing undifferentiated subtype. The tumor cells form irregular islands intimately intermingled with inflammatory infiltrates. The tumor cells are relatively large with scant, lightly eosinophilic cytoplasm and indistinct cell borders, vesicular nuclei, and prominent nucleoli. Keratin formation is difficult to be identified on routine H&E-stained section. (e) Nasopharyngeal carcinoma, metastatic to the neck lymph node. Immunohistochemical stains with pan-cytokeratin AE1/AE3 antibody shows uniform strong reactivity in the tumor cells (brown color), but no staining in surrounding lymphocytes. (f) Nasopharyngeal carcinoma, metastatic to the neck lymph node. EBV in situ hybridization for EBER shows positive nuclear reaction in tumor cells (blue color)
Gross and microscopic features: Nasopharyngeal carcinoma usually arises from the lateral wall of the nasopharynx, especially the fossa of Rosenmüller. Grossly, it may form a smooth bulge or nodule in the mucosa, with or without surface ulceration. Sometimes there is no visible lesion found, and the diagnosis is made by random biopsy in suspicious areas [19].
Microscopically, there are three distinctive subtypes: keratinizing squamous cell carcinoma (SCC), non-keratinizing carcinoma, and basaloid SCC. Keratinizing SCC resembles the usual well-differentiated SCC arising in other locations. There is obvious squamous differentiation with intercellular bridges and abundant keratinization at the light microscopic level. This type of tumor often occurs in an older age group and may not be associated with EBV infection.
Non-keratinizing carcinoma, which represents the large majority of nasopharygeal carcinoma, is associated with EBV infection in practically all cases. It may be subclassified into differentiated and undifferentiated types, the latter accounting for majority of cases. This subclassification is generally considered unnecessary, because it lacks clinical significance and lesions may show heterogeneity in different areas in the same biopsy or in different biopsies taken at different time intervals [20, 21]. However, a recent study showed worse prognosis with differentiated histology [22].
Tumor cells in non-keratinizing carcinoma typically form solid sheets or irregular islands intimately intermingled with variable numbers of inflammatory infiltrates rich in lymphocytes. Sometimes the lymphocytes may dominate the entire lesion and obscure the epithelial nature of the cells, mimicking a lymphoma. Undifferentiated subtype cells exhibit a syncytial appearance with scant, lightly eosinophilic cytoplasm and indistinct cell borders, vesicular nuclei, and prominent nucleoli (Fig. 7.1d), whereas differentiated subtype cells demonstrate some level of cellular stratification or pavement arrangement, often described as resembling transitional cell carcinoma of the bladder. Tumors may have focal or extensive spindle cell morphology or form papillary fronds. Other occasional finds include scattered spherical amyloid globules [23], epithelioid granulomas [24], and prominent infiltration by eosinophils and plasma cells [25].
The basaloid variant is the rarest type of NPC, as only a few cases are reported in the literature [26, 27]. This type of tumor is morphologically identical to neoplasms occurring in other head and neck sites but show a lower clinical aggressiveness. The tumors were composed of two types of cells, basaloid and squamous cells. The basaloid cells are small with hyperchromatic nuclei, inconspicuous nucleoli, and scant cytoplasm. The cells form closely packed solid sheets, irregular islands, nests, or cords, occasionally with peripheral palisading. A component of conventional SCC foci is invariably present in the basaloid variant, and the junction between the squamous and basaloid cells may be abrupt. Careful examination of the entire specimen to find the areas with conventional SCC may aid diagnosis. Another feature of basaloid SCC is the presence of stromal hyalinization with small cystic spaces containing PAS and Alcian blue-positive material. Comedo-type necrosis is frequent.
Immunohistochemistry and other special stains: The tumor cells show uniform strong reactivity for pan-cytokeratin AE1/AE3, cytokeratin 5/6, and p63, focal or weak reactivity for low-molecular-weight cytokeratins and EMA, and no reactivity for cytokeratins 7 and 20 [28–30] (Fig. 7.1e). Most tumors, especially non-keratinizing carcinoma, show a positive nuclear reaction for EBV-encoded early RNA (EBER) by in situ hybridization [31–33] (Fig. 7.1f). High-level expression of ERCC1 may be associated with more aggressive clinical behavior [22].
Molecular diagnostic features and cytogenetics: Although of no diagnostic value, rearrangement and deletion on chromosome 3 have been consistently noted in nasopharyngeal carcinoma [34–37].
Prognostic features: The mainstay of treatment for nasopharyngeal carcinoma is concomitant chemotherapy and radiation, with or without neoadjuvant chemotherapy. Progressive improvement has been reported both from endemic and non-endemic areas. The outcome in pediatric patients is usually better than that of adults, and the presence of metastatic disease in cervical lymph nodes at diagnosis apparently does not adversely affect prognosis. Development of therapy-related complications including second malignancy is of special concern in long-term survivors [16, 38–41].
NUT Midline Carcinoma
Definition: NUT midline carcinoma is a rare aggressive subset of poorly differentiated SCC, genetically defined by rearrangement of the Nuclear Protein in Testis [42] gene at chromosome 15q13 [43].
Clinical features and epidemiology: NMC is a newly described carcinoma commonly occurring in children and young adults, with a median age of 16 years (range 0.1–78) at the time of diagnosis [5, 44–46]. The majority of tumors arise in the midline structures in head and neck or in the thorax, and nearly one-half of the cases present with either lymph node or distant metastases [45, 46]. Rarely, the tumors arise in salivary glands, liver and pancreas, testis, and bladder [44, 47–50]. None of the cases tested to date have been associated with EBV or HPV infection [51].
Imaging features: Imaging appearance of NUT midline carcinoma is nonspecific, and very few case reports are present in the imaging literature. Imaging features include heterogeneous low density on CT and heterogeneous but predominant T1 hypointensity and T2 hyperintensity on MRI with heterogeneous enhancement [52]. Metastasis may occur in any part of the body and metastatic intraspinal and intracranial involvement have been described [52, 53]. The intracranial lesion may demonstrate restricted diffusion [52]. Intralesional calcification has also been reported [52]. NMC has been shown to be avid on PET imaging [52, 54].
Molecular genetics: NUT midline carcinoma is a genetically defined neoplasm caused by chromosomal rearrangement of the gene encoding NUT at 15q13. Approximately two-thirds have a translocation t(15;19)(q13;p13.1) involving NUT and BRD4, resulting in a BRD4–NUT fusion oncogene [43, 55]. Less common tumors have a different rearrangement involving NUT, of which t(9;15)(q34.2;q13) with BRD3–NUT fusion gene is the most common variant [55]. There is no significant association between translocation type (BRD4–NUT, BRD3–NUT, or NUT variant) and outcome, although some studies suggest that NUT-variant cancers may be associated with longer survival [44, 46].
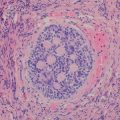
Gross and microscopic features: The histology of NUT midline carcinoma mimics many tumors arising in these locations. Most tumors have poorly differentiated carcinoma morphology with uniform round, oval, or spindle-shaped tumor cells arranged in sheets, islands, or ribbons, with or without desmoplastic stroma. The tumor cells often have high nuclear-to-cytoplasm ratios with inconspicuous cytoplasm, dense chromatin, and absent nucleoli. Occasional cases show abrupt squamous differentiation with minimal keratinization (Fig. 7.2). Sometimes neuroendocrine structures mimic neuroblastoma or Ewing’s sarcoma/primitive neuroectodermal tumor (PNET). Apparent chondroid differentiation has been described [47]. The tumor often shows brisk mitoses, apoptosis, and focal necrosis.
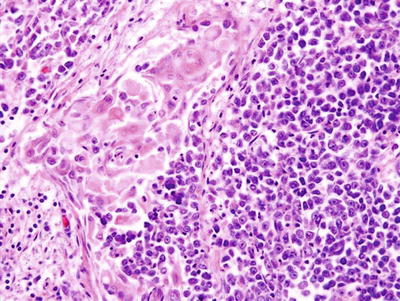
Fig. 7.2
NUT midline carcinoma. The tumor is typically composed of poorly differentiated, uniform round, oval, or spindle-shaped tumor cells (right half of the picture) with occasional abrupt squamous differentiation with minimal keratinization (left half of the picture)
NUT midline carcinoma is often confused with similar, poorly differentiated carcinoma features, such as poorly differentiated SCC, Ewing’s sarcoma/PNET, nasopharyngeal carcinoma, and pancreatoblastoma [51, 56].
Immunohistochemistry and other special stains: Immunohisto- chemical staining with a monoclonal antibody against NUT has been proven highly sensitive and specific for the diagnosis of NUT midline carcinoma [57]. In addition, the tumors express cytokeratin and p63, in keeping with squamous cell differentiation [56]. Occasionally, there is negative staining for keratins including a pan-keratin cocktail, Cam5.2, and/or AE1/AE3 [58]. The tumor is in general negative for sarcoma, melanoma, and lymphoma markers.
Molecular diagnostic features and cytogenetics: NUT midline carcinoma can be diagnosed by conventional cytogenetics with characteristic t(15;19). Since the discovery of NUT rearrangement and its partner genes, reverse transcriptase (RT)-PCR and fluorescence in situ hybridization (FISH) have been used for the diagnoses but are largely replaced by NUT immunohistochemistry (see above). Nonetheless, they remain the gold standard for confirming the diagnosis.
Prognostic features: All NUT midline carcinomas show aggressive behavior with early locoregional invasion and distant metastases. They are often initially responsive to chemotherapy and radiation but invariably recur and do not respond to subsequent therapeutic interventions. The overall survival at 1 and 2 years after diagnosis has been 30 and 19 %, respectively, and the average survival is less than 1 year [46].
Esthesioneuroblastoma (Olfactory Neuroblastoma)
Definition: Esthesioneuroblastoma, also called olfactory neuroblastoma, is a malignant neuroendocrine tumor arising from the olfactory mucosa of sinonasal tract and frequently invading into the orbits and skull base.
Clinical features and epidemiology: Esthesioneuroblastoma is an uncommon tumor, accounting for approximately 3–6 % of all sinonasal malignancy [59, 60]. It usually occurs in adults between the ages of 40 and 70 years (mean 53) [61] but is rare in children; only 10 % of reported cases in English literature are in the pediatric population [62]. Most pediatric patients are adolescents, with slight male predominance (1.5:1). Patients usually present with a nasopharyngeal polypoid mass that may cause unilateral nasal obstruction, local swelling, facial pain, and recurrent epistaxis. The tumor may protrude into the orbit and cause proptosis, ophthalmoplegia, and even visual loss, or extend via the cribriform plate into the cranium. The resultant frontal lobe lesion mimics a brain tumor [63, 64]. Occasional patients may present with Cushing syndrome or hyponatremia due to ectopic ACTH or ADH production [6, 65, 66]. Pediatric esthesioneuroblastoma seems to have a more aggressive presentation than in adults [67]. There is no evidence of EBV infection [68].
Imaging features: Esthesioneuroblastoma and its imaging characteristics have been well described in the adult literature. Because these are rare tumors in the pediatric population, there is paucity of literature focusing on imaging of this tumor in children. Typically, esthesioneuroblastoma demonstrates a large aggressive-appearing nasal mass with common extension into the paranasal sinuses and erosion of the cribriform plate and orbital wall, with intracranial and/or orbital extension (Fig. 7.3a). On CT, they are usually homogenous, enhancing masses which cause bone remodeling. As mentioned, they can involve the nasal cavity and the paranasal sinuses. On MRI, esthesioneuroblastoma has intermediate signal on T1 and T2 (Fig. 7.3a–c) and enhances with gadolinium. In tumors with intracranial extension, peripheral cysts can be present at the margins of the intracranial mass and are helpful in suggesting the diagnosis of esthesioneuroblastoma [69]. Erosion of the paranasal sinuses is common. In contrast to neuroblastoma in other locations, metaiodobenzylguanidine (MIBG) scans have been shown to be negative in a series of patients with esthesioneuroblastoma [67].
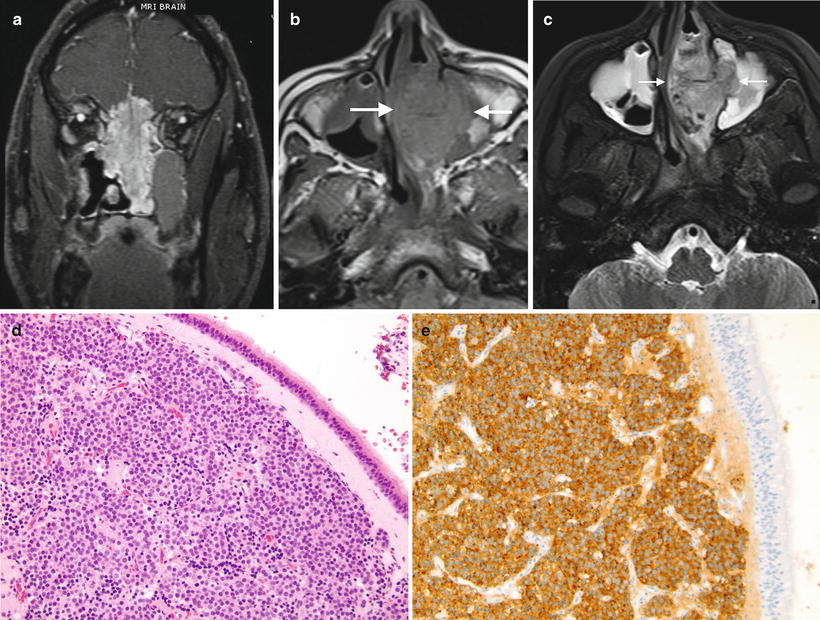
Fig. 7.3
(a) Esthesioneuroblastoma. Coronal T1W postcontrast MRI image in an adult patient demonstrates the common appearance of esthesioneuroblastoma, with a large nasal mass extending into the ethmoid sinuses and causing erosion of the cribriform plate, with orbital and intracranial extension. Image courtesy of Dr. Nicholas Weisman, Yale New Haven Hospital. (b, c) 14-year-old girl with left nasal esthesioneuroblastoma (b) Non-contrast-enhanced T1W axial MRI shows that the tumor is isointense to muscle and infiltrates the left maxillary sinus. (c) Axial T2W MRI shows the tumor to be slightly hyperintense to muscle. Mucoperiosteal thickening of both maxillary sinuses is present. Images courtesy of Dr. Beth McCarville, St Jude Children’s Research Hospital, Memphis, TN. (d) Esthesioneuroblastoma. The tumor is located in the subepithelial region of the nasal mucosa. It is composed of nests of primitive small round blue cells invested by fibrovascular stroma. (e) Esthesioneuroblastoma. The tumor cells show strong expression of synaptophysin demonstrated by immunohistochemical stain
Gross and microscopic features: Esthesioneuroblastoma is a small round blue cell tumor that resembles neuroblastoma arising from adrenal gland or sympathetic chain. Grossly, tumors form a red-gray, highly vascularized, polypoid mass, commonly located in the roof of the nasal fossa. Sizes range from <1 cm up to large masses involving the nasal cavity and intracranial region.
Microscopically, esthesioneuroblastoma has a lobular architecture composed of nests of cells invested by fibrovascular stroma. The cellular components consist of small- to medium-sized primitive cells with high nuclear-to-cytoplasmic ratio, uniform round nuclei with dispersed coarse “salt and pepper”-appearing chromatin, and inconspicuous nucleoli (Fig. 7.3d). The background often has a neurofibrillary appearance with occasional Homer Wright-type rosettes, which are nearly pathognomonic in the nasal cavity when containing true neuropil [70]. The stroma between the tumor lobules is fibrotic and often contains a prominent vascular proliferation that may obscure the histology of the underlying neoplastic process [71]. Uncommon findings include occasional calcification, melanin-containing cells, ganglion cells, and divergent differentiation with rhabdomyoblasts or epithelial islands [70]. Ganglioneuroblastic transformation after chemotherapy has been reported [72].
Immunohistochemistry and other special stains: In keeping with their neuroblastic differentiation, esthesioneuroblastomas show strong immunoreactivity for neuroendocrine marks including synaptophysin (Fig. 7.3e), chromogranin, CD56, neuron-specific enolase [73], neurofilament protein, and calretinin [74]. S-100 protein is usually expressed in the sustentacular cells at the periphery of the tumor lobules. A few esthesioneuroblastomas may focally express cytokeratin or p63, but they usually show negative immunoreaction to EMA [75, 76]. In general, esthesioneuroblastomas show negative immunoreactivity to HMB45, desmin, myogenin, CD99, and leukocyte common antigen, unless heterologous elements are present (see above).
Molecular diagnostic features and cytogenetics: Molecular genetic data is sparse for esthesioneuroblastoma. Several recent studies have demonstrated extremely complex genetic changes, but none has been sufficiently recurrent to be helpful in diagnosis [77–79]. The constant absence of t(11;22) or EWS rearrangement [80–82] indicates that esthesioneuroblastoma is not related to Ewing’s sarcoma.
Prognostic features: In pediatrics, the first line of treatment for esthesioneuroblastoma is a combination of chemotherapy and radiotherapy. The most important predictors of outcome are tumor stage, treatment modality, lymph node status, and age at diagnosis. The prognosis is better in children in comparison to adult patients [67], with overall 5-year survival rates of 45.6 % in adults and 88.9 % in children [61, 67]. Lymph node metastasis, which occurs in approximately 23 % of the cases, has been considered as an important prognostic factor; therefore an elective neck treatment has been recommended [83–85].
Sarcomas Affecting the Nasal Area
Sarcomas arising in nasal area are rare and account for no more than 5 to 10 % of all malignant tumors in head and neck region. Compared to adults, children have a somewhat higher proportion of sarcomas in the head and neck and about 40 % of all rhabdomyosarcoma arise in the region. Among head and neck rhabdomyosarcoma, 35–50 % arise in the sinonasal and nasopharyneal areas [86, 87]. The majority in the sinonasal region are alveolar type and have worse prognosis than tumors in other head and neck areas [86]. Other rare sarcomas in the sinonasal region include fibrosarcoma, leiomyosarcoma, malignant fibrous histiocytoma, angiosarcoma, and malignant peripheral nerve sheath tumor (MPNST). The morphology, immunohistochemistry, and molecular features of sinonasal sarcomas are similar to the same tumors occurring in other parts of the body.
Imaging features: Rhabdomyosarcoma in the paranasal regions is often advanced and locally invasive. Imaging usually demonstrates a poorly defined enhancing mass commonly with bony destruction and intracranial extension (Fig. 7.4). CT is useful to demonstrate the degree of bony erosion. When in the nasal soft tissues, rhabdomyosarcoma may present as a relatively small soft-tissue mass without bony destruction. The absence of bony erosion is also common in rhabdomyosarcoma in other head and neck regions. MRI is especially useful in the evaluation of intracranial extension as well as on follow-up of these tumors.
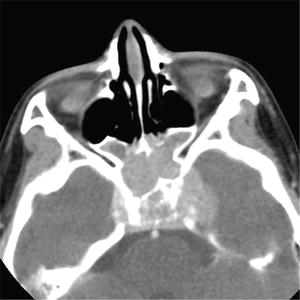
Fig. 7.4
Rhabdomyosarcoma. Contrast-enhanced CT image of a young girl shows a large mass centered in the sphenoid sinus with intracranial extension and bony destruction of the skull base
Oral Cavity and Salivary Gland Carcinomas in Children
Salivary glands are exocrine organs and comprise three paired major glands (the parotid, submandibular, and sublingual), and numerous minor widely distributed throughout the mouth and oropharynx. The global annual incidence of malignant salivary gland tumors ranges from 0.4 to 0.9 cases per 100,000 population [88, 89]. The peak incidence of salivary gland carcinomas is in the sixth and seventh decades [90, 91]. From 1973 to 2006, Sultan et al. identified 12,834 cases of salivary gland carcinomas reported to the Surveillance, Epidemiology, and End Results (SEER) database, of which only 263 cases (2 %) occurred in children and adolescents (<20 years) [92]. Mucoepidermoid carcinoma is the most common primary salivary gland malignancy in both adults and children and often presents as a secondary cancer in pediatric patients with a history of nonsalivary cancer. Sialoblastoma is unique to pediatrics as most tumors present congenitally or during early infancy. Acinar cell carcinoma and adenoid cystic carcinomas are very rare in the pediatric population and may act similarly to those occurring in adults [93].
Mucoepidermoid Carcinoma
Definition: Mucoepidermoid carcinoma is a malignant glandular epithelial tumor arising from the large ducts of both major and minor salivary glands. It is principally composed of three types of cells, mucinous, squamous, and intermediate, that display columnar, clear cell and oncocytoid features.
Clinical features and epidemiology: Although rare, mucoepidermoid carcinoma is the most common malignant salivary gland tumor in childhood. Many pediatric patients have a history of chemotherapy or radiation for a nonsalivary malignancy [94]. Most tumors arise in the major salivary glands, predominantly in the parotid. Occasionally, the tumors occur in minor salivary glands, most commonly in the palate. Rarely, the tumor may occur in the trachea, nasal cavity, and other locations [95]. Most patients present with an isolated, painless, and slow-growing mass. The clinical and pathologic features are similar in pediatric and adult populations.
Imaging features: Diagnosis of mucoepidermoid carcinoma may be difficult based on imaging alone, and tissue diagnosis is usually needed. On computed tomography, these tumors frequently appear as well-defined masses with moderate enhancement (Fig. 7.5a). Magnetic resonance can be useful in evaluating soft-tissue masses in the salivary glands, since it has better tissue resolution. The mass is usually hyperintense to the adjacent gland on T2-weighted images, and however hypointense compared to the adjacent lymph nodes [96]. Differential diagnosis includes vascular malformations as well as pleomorphic adenoma, the most common salivary gland tumor in children. Pleomorphic adenomas tend to have higher signal intensity on T2-weighted MR images compared to mucoepidermoid carcinoma [97]. Other malignancies such as adenoid cystic carcinoma, acinic cell carcinoma, and lymphoma may have similar imaging findings to mucoepidermoid carcinoma. Imaging features of atypical infection may also overlap.
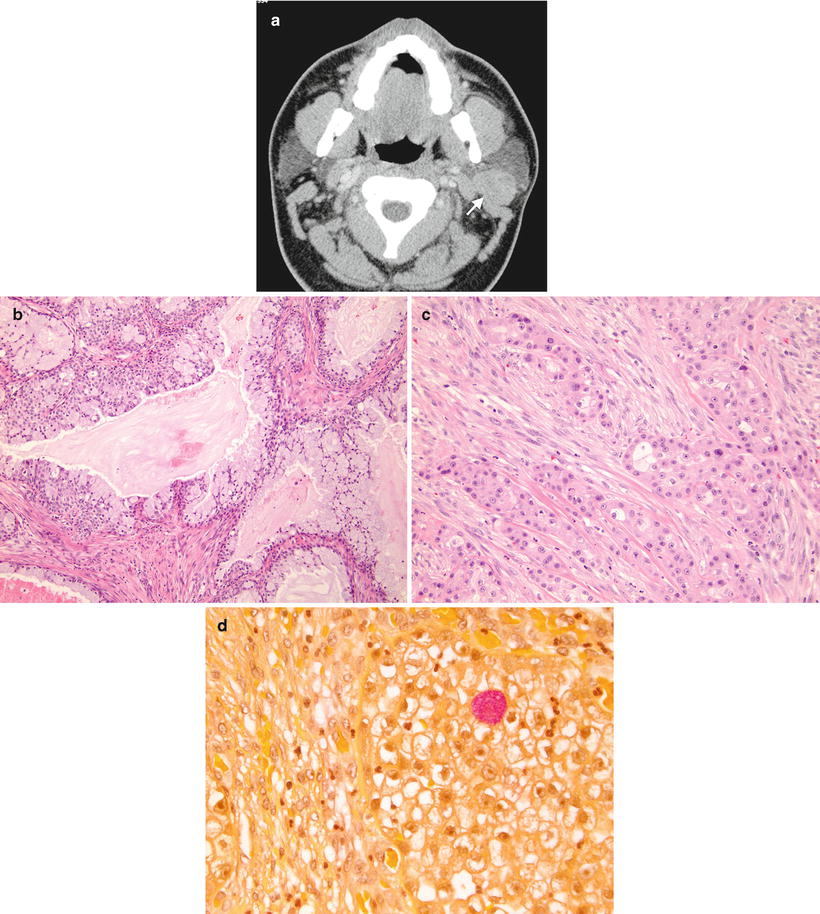
Fig. 7.5
(a) Mucoepidermoid carcinoma, left parotid gland. Postcontrast CT image demonstrates a moderately enhancing, well-circumscribed mass within the posterior aspect of the left parotid gland (arrow). Biopsy confirmed mucoepidermoid carcinoma. (b) Mucoepidermoid carcinoma, low grade. The tumor consists predominately of well-differentiated mucin-producing cells with well-formed glandular structures and cystic spaces. (c) Mucoepidermoid carcinoma, high grade. The tumor is composed of solid sheets of squamous and intermediate cells with minimal mucinous cells. (d) Mucoepidermoid carcinoma, high grade. The mucus cell contains abundant foamy cytoplasm that stains positively with mucicarmine special stain (pink color)
Gross and microscopic features: Grossly, mucoepidermoid carcinoma is firm, often cystic, and tan with well-defined or infiltrated margin. Microscopically, it is principally composed of three types of cells: mucin producing, squamous (epidermoid), and intermediate. Mucus cells vary in shape and contain abundant foamy cytoplasm that stains positively with mucin stains. Squamous cells are usually polygonal shaped and show intercellular bridges and occasional keratinization. Intermediate cells are basaloid in appearance and believed to be able to differentiate into the other two cell types [98]. Some tumors also show variable numbers of clear cells or lymphoid infiltrates.
Mucoepidermoid carcinoma can be divided into low-, intermediate-, and high-grade types. Low-grade tumors consist of well-differentiated mucin-producing cells which produce well-formed glandular structures or cystic spaces (Fig. 7.5b). High-grade tumors have a more cellular appearance and are composed largely of squamous and intermediate cells with minimal mucinous cells (Fig. 7.5c). The intermediate-grade tumor usually has more intermediate and squamous cells than the low-grade lesion, with occasional cysts and intracystic proliferation of intermediate or squamous cells. There is usually no marked nuclear atypia, brisk mitosis, or extensive necrosis in any grade of this tumor.
Immunohistochemistry and other special stains: Sialomucin content of mucin-producing mucoepidermoid carcinoma cells is demonstrated by mucicarmine or Alcian blue staining (Fig.7.5d). This special stain is particularly useful in high-grade tumors, in which the mucin-producing cells are usually sparse. Cytokeratin, especially high-molecular cytokeratin, and p63 may be used to identify squamous cells [99].
Molecular diagnostic features and cytogenetics: A nonrandom t(11;19) reciprocal translocation with a CRTC1-MAML2 fusion oncogene is frequently identified in mucoepidermoid carcinoma by conventional cytogenetics, FISH, and reverse transcription polymerase chain reaction [99]. However, this translocation may be found in some benign salivary tumors such as Warthin’s tumour and clear cell hidradenoma, so that interpretation must be performed in the context of histopathology.
Prognostic features: Treatment of mucoepidermoid carcinoma includes surgery or radiation, or both. The most important prognostic factors are the tumor grade and stage. Tumors in children have a better prognosis. One study showed that the 5-year survival rate in pediatric mucoepidermoid carcinoma was 93.7 %, and survival rate did not differ in patients with secondary tumors [94]. Mucoepidermoid carcinoma with CRTC1–MAML2 may have a better prognosis [100].
Sialoblastoma
Definition: Sialoblastoma is a rare, malignant epithelial salivary gland neoplasm that usually present at birth and recapitulates the primitive salivary anlage [101].
Clinical features and epidemiology: Most sialoblastomas present congenitally or during early infancy. Some patients may be diagnosed by prenatal ultrasound. Occasional children may be diagnosed after the age of 2 years. Most patients present with respiratory difficulty shortly after birth. The male-to-female ratio is 2:1. The tumor commonly arises in the parotid or submandibular gland. Originally considered as a benign neoplasm, sialoblastomas have documented locoregional recurrence and distant metastases and so are included among malignant epithelial salivary gland neoplasms. The simultaneous occurrence of hepatoblastoma has been reported [73, 102, 103].
Imaging features: Very few case reports describe the imaging features of sialoblastoma. CT shows a mass which is usually hypoattenuating compared to adjacent muscle. On MR, this tumor has been demonstrated to have isointense signal to muscle on T1-weighted images and an intermediate-to-high signal intensity on T2-weighted images [21]. On contrast-enhanced T1-weighted images sialoblastoma usually demonstrates heterogeneous contrast enhancement [21]. Intralesional hemorrhage and necrosis have also been described [104].
Gross and microscopic features: Grossly, sialoblastoma is a firm, smooth, polypoid mass measuring 2–7 cm in greatest dimension [105]. Microscopically, tumors have a biphasic pattern: basaloid epithelial cells that form ductules or bud-like structures and solid organoid nests, and relatively hypocellular spindle cell stroma (Fig. 7.6). The basaloid epithelial cells have scanty cytoplasm, round-to-oval nuclei, single or few nucleoli, and relatively fine chromatin pattern. More mature cuboidal epithelial cells with squamous differentiation can be seen, and some form solid squamous nests or duct structures resembling sialometaplasia. The mitotic rate within sialoblastomas is highly variable and may increase with subsequent recurrences [106]. Significant necrosis and marked pleomorphism are uncommon in this tumor.
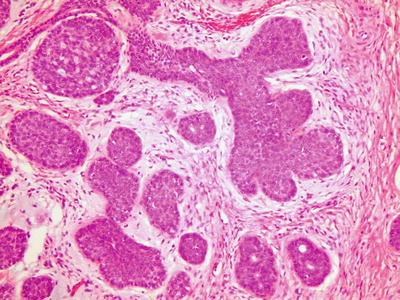
Fig. 7.6
Sialoblastoma. The tumor has a biphasic pattern: basaloid epithelial cells with squamous features that form solid organoid nests with focal ductule-like structures, and relatively hypocellular spindle cell stroma (picture courtesy Deborah Perry, M.D., Children’s Hospital & Medical Center, University of Nebraska Medical Center College of Medicine)
Immunohistochemistry and other special stains: Sialoblastomas show diffuse and widespread reactivity for salivary gland amylase. The epithelial components express p63, cytokeratin, EMA, CK5/6, CK7, and S-100. There is no expression for CK20. The stroma spindle cells show focal reactivity to smooth muscle actin [101, 107].
Molecular diagnostic features and cytogenetics: One case report of a sialoblastoma showed clonal chromosome aberrations with a complex karyotype [106].
Prognostic features: Surgery is the treatment of choice. There is limited prognostic data on this tumor due to its rarity. One study has suggested that the presence of anaplastic basaloid tumor cells, minimal stroma, and broad pushing to infiltrative periphery may be related to more aggressive behavior [105].
HPV-Related Carcinoma
HPV has become a considerable concern in cancer epidemiology and is linked to carcinomas of the head and neck and male and female genitalia. In children, HPV-associated neoplasms primarily arise in the context of congenital infections presumably acquired around the time of labor and delivery. These lesions primarily affect the mouth, pharynx, and larynx and may give rise to solitary or multiple papillomas. Juvenile oropharyngeal papillomas usually are benign lesions comprising a layer of HPV-infected stratified squamous epithelium overlying a fibrous core. They show variable degrees of dysplasia and atypia, as well as koilocytosis. HPV types 6 and 11 predominate in these lesions [108, 109]. The tumors may be aspirated into the lungs and cause bronchial based lesions. Rarely they give rise to well-differentiated squamous cell carcinomas, with cervical nodal metastasis [110].
A rising health care concern is the increasing incidence of oral cancers among younger nonsmoking adults, primarily as a result of sexual exposures to oncogenic HPV types 16 and 18 [3, 111]. Sites affected include the tongue and tonsils [112, 113]. Although adolescents are reportedly not affected by metastatic cancers, we have seen oral in situ carcinomas in our dental clinic.
Imaging features: Masses within the oral cavity and larynx are frequently diagnosed by direct visualization and laryngoscopy. Imaging plays an important role in evaluation of the submucosal involvement, involvement of deep soft tissues and bone, as well as in evaluation for distant metastases [118]. Squamous papillomas can be solitary or multiple. The multiple form is also known as juvenile laryngotracheal papillomatosis. Computed tomography demonstrates nodules within the larynx and tracheobronchial tree and lungs. CT is important in evaluating the extent of these lesions and degree of tracheal and lung involvement. The possibility of malignant transformation of papilloma makes follow-up by CT important in these patients [114]. Squamous cell carcinoma of the oral cavity may appear as homogeneous or heterogeneous masses with variable contrast enhancement. Contrast enhancement may predominate along its margins [115]. On MRI, SCC has predominantly low-to-intermediate T1 signal and intermediate-to-high T2 signal. Delineation of soft tissue involvement is better demonstrated with MRI. A significant percentage of SCC of the oral cavity present with lymph node metastases.
Melanotic Neuroectodermal Tumor of Infancy
Clinical features and epidemiology: Melanotic neuroectodermal tumor of infancy is a rare neoplasm of neural crest origin that presents in the first year of life in 95 % of cases, typically involves the mandible or maxilla, and has similarities to neuroblastoma and Ewing’s sarcoma histologically but is genetically unique [116]. The tumor may present congenitally, and occasionally can involve sites outside of the jaw, such as the leptomeninges, genitourinary system, and the extremities. Like neuroblastoma, plasma and urine catecholamine metabolites may be elevated. Other names that have been applied to this tumor in the past are retinal anlage tumor and progonoma. Although the majority (greater than 90 %) behave in a benign fashion, there are recorded cases of metastases and fatal outcome [117].
Imaging features: About 70 % of MNTI arise in the maxilla, followed by 11 % in the skull and 6 % in the mandible. Skull lesions generally arise around the sutures with about 50 % occurring near the anterior fontanelle. Parenchymal brain involvement, when present, results from direct tumor extension, although there are reports of MNTI arising in the third ventricle and cerebellar vermis. Tumors generally compress adjacent structures rather than infiltrating them. In the mandible and maxilla they may cause tooth displacement, bone destruction, expansion, or remodeling.
Plain-film radiography: Initial radiographs may show a well-marginated, non-aggressive, radiolucent lesion with or without irregular margins in the skull or facial bones [118, 119]. When tumors arise in the mandible or maxilla, the differential includes developmental cysts, odontogenic lesions, infection, fibrous dysplasia, and vascular malformations. In contrast to benign lesions, MTNI grow rapidly resulting in bone destruction, a finding that can narrow the differential to more aggressive lesions. Occasionally a faint spiculated or sunburst appearance may be present [120].
Computed tomography: CT can accurately define the extent of the lesion and aid in surgical planning [119]. Tumors appear expansile with a well-defined soft-tissue component that may be slightly hyperdense on non-contrast-enhanced images due to melanin content. The tumors generally enhance after administration of contrast material. Maxillary lesions may cause “floating teeth” while calvarial lesions may show spiculation and hyperostosis [116, 120].
Magnetic resonance imaging (MRI): On MRI, maxillary and calvarial tumors may appear variably hyperintense on T1-weighted and hypointense on T2-weighted images depending on the amount of melanin within the tumor. The tumor may also contain areas that are hypointense on both T1- and T2-weighted images due to calcification and hyperostosis (Fig. 7.7a–c). After administration of contrast material, MTNI typically demonstrate intense enhancement in non-calcified areas. On diffusion-weighted MRI, cellular areas of tumor will demonstrate restricted diffusion. Sinovenous involvement, which is sometimes associated with this tumor, is best assessed with magnetic resonance angiography [118, 119].
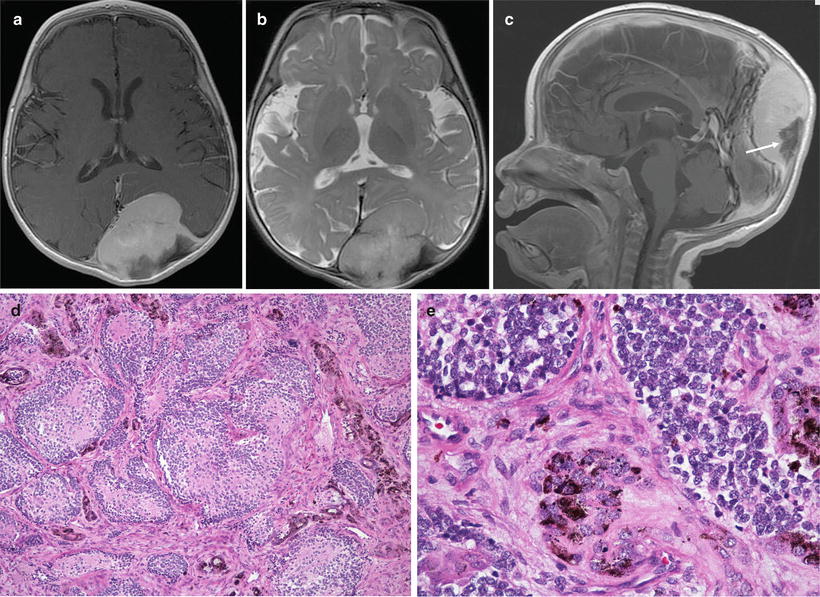
Fig. 7.7
(a–e) Melanotic neuroectodermal tumor of infancy arising in a 5-month-old male. (a) Axial T1W postcontrast MRI image shows the extra axial tumor to be homogenously enhancing. (b) Axial T2W MRI image shows the tumor to be predominantly isointense to white matter, and causing mass effect on the underlying parenchyma. (c) Sagittal postcontrast T1W MRI image shows contrast enhancement of the tumor except in the area of spiculated hyperostosis, a feature typical of MNTI. (d) Nested, neuroblastoma-like pattern with pigmented cells in the peripheral fibroblastic stroma. (e) Higher power view of D, demonstrating coarse melanin granules at the periphery of the small cell nests
Gross and microscopic features: Ranging in size from 1 to 10 cm, these tumors are gray-black and firm to palpation, reflecting the presence of pigment and a prominent stromal component. Histologically, nests of tumor cells are surrounded by dense fibrous stroma (Fig. 7.7d, e). The cellular nests are composed of primitive small neuroblast-like cells with hyperchromatic nuclei and little cytoplasm. These cells may form neuroblastic type rosettes, and in some cases may be associated with neuropil. Mitoses are absent to rare. Larger, melanin-containing cells are present, either interspersed with the neuroblast-like cells or present at the periphery of the nests [117, 121].
Immunohistochemical and other special stains: The large cells of melanotic neuroectodermal tumors stain immunohistochemically with melan-A, HMB-45, vimentin, cytokeratin AE1/AE3, and epithelial membrane antigen but are negative with S-100. Cytoplasmic melanin can be demonstrated with the Fontana stain. The neuroblast-like cells stain with neural markers such as NSE and CD56, and have variable staining for chromogranin, synaptophysin, and GFAP. Occasional cases have shown positivity with desmin and muscle-specific actin, and may even show focal myogenin positivity [121, 122]. CD99 is generally not expressed, although one of the eight cases in one study had membranous staining [123]. Cases subjected to electron microscopic examination have shown the small cells to share the ultrastructural features of dense core neurosecretory granules and dendritic processes with neuroblastoma, whereas the larger cells contain melanosomes [124].
Molecular diagnostic features and cytogenetics: Molecular genetic data is sparse for this rare neoplasm, and the diagnosis rests largely upon its biphasic light microscopic appearance and polyphenotypic immunohistochemical profile. Unlike Ewing’s sarcoma, these tumors lack a t(11;22) translocation [116], and unlike neuroblastoma none of these tumors have shown evidence of MYCN amplification or 1p deletion [124].
Lymphoma/Leukemia of the Oral Cavity
Pediatric lymphomas have a well-known tendency to first appear as enlarged cervical lymph nodes or neck masses and are more extensively discussed in the chapter on hematopoietic and lymphoid tumors. Of particular note, however, in a discussion of pediatric head and neck tumors is the tendency of both Burkitt lymphomas and monoblastic leukemias to present as jaw masses. Gnathic Burkitt lymphomas have been well described in the original literature of the endemic, EBV-associated form of the disease discovered in sub-Saharan Africa by Burkitt in 1952 [125]. For a fascinating history of the association between Burkitt lymphoma and EBV, the reader is referred to reference [126]. Outside of this setting, Burkitt lymphoma of the jaw is distinctly rare [127].
The head and neck, particularly the oral cavity and jaw, comprise a relatively common site for myeloid sarcomas. Monoblastic leukemias of the jaw tend to occur in young patients and are usually associated with 11p23 translocations involving the MLL gene [128]. Other leukemias may also present in this manner [129], and the leukemia may be first discovered by histological examination of soft tissues associated with an extracted tooth. The gingival tissues appear to be one of the preferred sites for extramedullary leukemias, which may present prior to leukemic manifestations of the disease.
Imaging features: Burkitt lymphoma in the head and neck typically affects the mandible or maxilla. On radiographs and CT, these appear as poorly defined lytic lesions with displacement of tooth buds [130]. The differential diagnosis for a lytic lesion involving the mandible/maxilla is however broad and include more common entities such as infection, benign cysts, Langerhans cell histiocytosis, and sarcomas.
Sarcomas of the Oral Cavity/Salivary Glands
A number of pediatric sarcomas show a predilection for the head and neck and have a particular propensity to arise in or near the mouth or salivary glands. The most common by far is embryonal rhabdomyosarcoma, which can affect soft tissues adjacent to the parotid gland [121]. Diagnosis may be accomplished by fine-needle aspiration [131]. Rarely, alveolar rhabdomyosarcoma also occurs in these regions [131, 132], but confirmation of fusion status via FISH, RT-PCR, or karyotyping is advisable to avoid over-treatment. Following radiation and chemotherapy, 5-year overall survival of parotid region rhabdomyosarcomas approaches 85 % [121].
Among pediatric non-rhabdomyosarcomatous soft-tissue sarcomas (NRSTS), epithelioid sarcoma [133], alveolar soft part sarcoma [134, 135], myofibrosarcoma [136], and synovial sarcoma [137, 138] share a propensity to occur in the head and neck, particularly in and about the oral cavity. The prognosis of these lesions generally depends on the adequacy of excision. Synovial sarcomas respond well to chemotherapy [139, 140].
Among bony sarcomas, osteosarcomas may occur in the jaw, where they appear to have clinicopathological characteristics distinct from those arising in extremities [141]. Most occur in the mandible. In children, they generally show an osteoblastic morphology and tend to be large and of high grade [54]. In spite of these features, they have a better outcome than extremity osteosarcomas [54].
Imaging features: Rhabdomyosarcomas in the head and neck usually present as a soft-tissue mass which may cause bone destruction or remodeling. The absence of bone involvement is fairly common, however, for smaller lesions. CT is the modality of choice to evaluate for bony involvement. On MRI the mass tends to be hypointense on T1, and of low, intermediate, or high signal intensity on T2 depending on the cellularity. Contrast enhancement is variable; however, postcontrast imaging is helpful for evaluation of intracranial extension of disease [4] (Fig. 7.8). The presence of associated metastatic lymphadenopathy is common at presentation and imaging of cervical lymph nodes should be included in the initial evaluation.
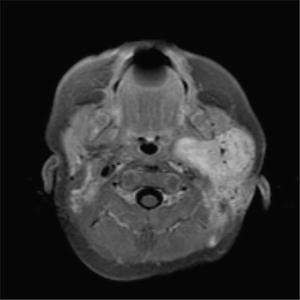
Fig 7.8
Rhabdomyosarcoma, left parotid gand. Axial T1W postcontrast MRI image demonstrates a large infiltrative enhancing mass involving the left parotid gland and extending into the deep cervical soft tissues and parapharyngeal space
Osteosarcomas of the head and neck most frequently affect the mandible and maxilla and frequently present with swelling and pain. On CT, they tend to have an aggressive osteolytic appearance with tumor matrix mineralization and a soft-tissue component [142, 143] (Fig. 7.9). An osteolytic lesion without tumor matrix mineralization is less common but can occur. CT is excellent in demonstrating tumor calcification, cortical involvement, and soft-tissue involvement as well as intramedullary extension. Periosteal reaction tends to be less pronounced than in long bone osteosarcoma [142]. MRI can be useful in evaluating adjacent structures and follow-up posttreatment. Cervical lymphadenopathy at presentation is uncommon [142].
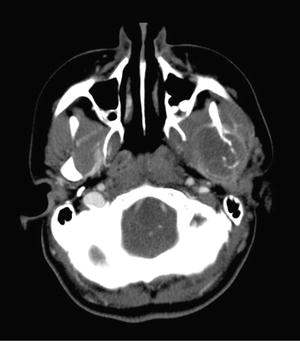
Fig 7.9
Osteosarcoma of the mandible. Contrast-enhanced CT image shows a fairly large, mostly rim-enhancing, mass involving the left mandibular ramus and condyle with intermixed calcific densities, bony destruction, and associated soft-tissue mass. The margins of the mass are difficult to distinguish from the surrounding musculature on CT and may be better delineated on MRI
Ear Tumors
Paragangliomas
Paragangliomas are more extensively discussed elsewhere in this text (see Tumors of Adrenal Gland and Extraadrenal Paraganglia). However, special mention must be made of the jugulotympanic paragangliomas that occur in the region of the ear or the glomus jugulare. These lesions may bulge against the tympanic membrane or into the jugular vein. Those in the ear cause problems with hearing, balance, or tinnitus, and those in the jugular region may extensively invade the base of the skull [144]. These lesions have a wide age range that includes children; fortunately they have a low metastatic rate, less than 5 % [145].
Imaging features: Paragangliomas (glomus tumors) may be confined to the middle ear (glomus tympanicum) or involve the region of the jugular bulb (glomus jugulare) with or without involvement of the middle ear due to destruction of the jugular plate (glomus jugulotympanicum). These are very rare in the pediatric population. On CT, the glomus jugulare or jugulotympanicum is a poorly defined soft-tissue mass with permeative bone destruction and avid diffuse contrast enhancement [146, 147]. A hyperintense mass on T2-weighted images is characteristic on MRI, with internal foci of hypointensity which represent high-flow vessels. Unenhanced T1-weighted images show their characteristic “salt-and-pepper” appearance. The “pepper” represents the hypointense foci caused by the signal void of large feeding vessels, whereas the “salt” is secondary to subacute hemorrhage or slow-flow vessels within the tumor [146, 147]. On angiography, this tumor is hypervascular, with rapid tumor blush and early draining veins. The glomus tympanicum has a characteristic appearance on CT which demonstrates a focal soft-tissue mass with flat base in the region of the cochlear promontory. The ossicular chain is usually spared and bone erosion is rarely present. These are frequently small tumors measuring up to 2 cm; however bone destruction may be present when the tumor is large. On T2-weighted images, these lesions may vary in signal intensity [146, 147]. Contrast-enhanced T1-weighted MR images usually demonstrate a strongly enhancing mass lesion within the middle ear.
Gross and microscopic features: Microscopically, jugulotympanic paragangliomas resemble those in other locations and contain epithelioid cells arranged in zellballen separated by rich vascular arcades and surrounded by sustentacular cells (Fig. 7.10a). They tend to be more vascular than other lesions, and they may show significant degrees of sclerosis. At times, they have a small cell, neuroblastoma-like configuration. Immunostains for neuroendocrine markers such as chromogranin and synaptophysin confirm the diagnosis. Immunostain for S-100 protein is positive in sustentacular cells, but negative in tumor cells (Fig. 7.10b). On occasion [148], rhomboid intracytoplasmic crystals that resemble those of alveolar soft part sarcoma are seen by electron microscopy.