Stage
Patients (%)
Characteristics
I
43
Limited to kidney
II
20
Extends beyond kidney but completely resected
III
21
Residual tumor confined to abdomen; nonhematogenous; abdominal/pelvic lymph nodes
Tumor rupture, spillage confined to flank
IV
11
Hematogenous (lung, liver, brain); extra-abdominal lymph nodes
V
5
Bilateral disease at diagnosis
Congenital Mesoblastic Nephroma
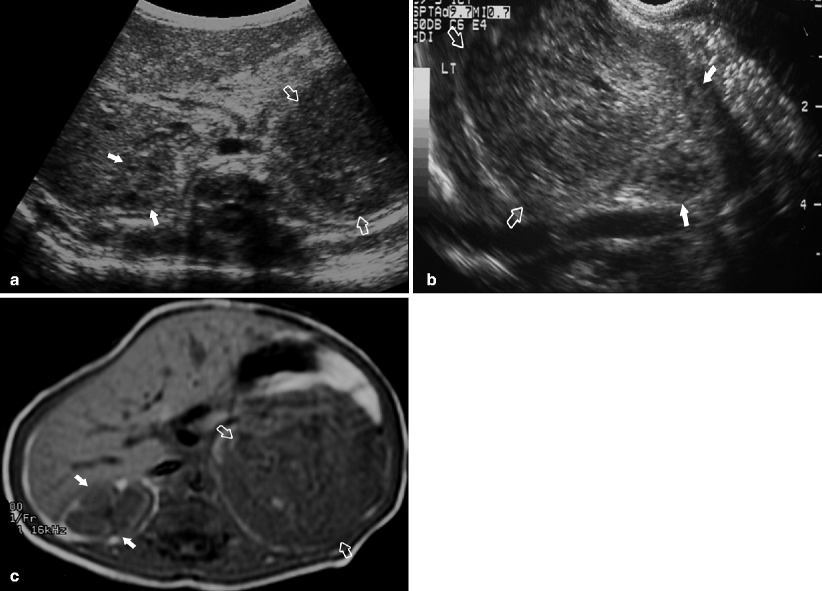
Fig. 10.1
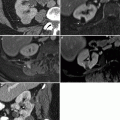
An abdominal mass was palpated on 4-week-old male during a routine physical examination. Transverse abdominal ultrasound (a) demonstrates normal right kidney (closed arrows) and large left renal mass (open arrows). On sagittal ultrasound images (b) the heterogeneous left upper pole mass (open arrows) and normal lower pole renal parenchyma (closed arrows) are seen. Axial post-gadolinium T1-weighted MR (c) shows a large vascular left renal mass (open arrows) and a normal right kidney (closed arrows). Surgical resection confirmed the suspected diagnosis of congenital mesoblastic nephroma (CMN)
CMN, also known as mesoblastic nephroma or fetal renal hamartoma, is the most common solid renal mass in infants under the age of 6 months [4]. The peak age of presentation is 1–3 months and 90 % of cases occur within the first year [5]. CMN is considered a low-risk renal tumor [2]. Although a palpable abdominal mass is the most common presentation in infants, some cases are detected on routine prenatal ultrasound examinations [2]. CMN is divided into classic and a more aggressive cellular subtype. Imaging in cases of the classic subtype demonstrates a large solid mass that may have peripheral enhancement. Necrosis/cystic changes, hemorrhage, focal enhancement, and infiltration of perinephric tissue can be seen in the cellular subtype [6]. Nephrectomy with wide margins may be the only treatment necessary. Although the tumor is generally considered as benign, local recurrence can occur in cases of incomplete resection. Metastases to lungs, brain, and bone are rare [2].
Nephroblastomatosis
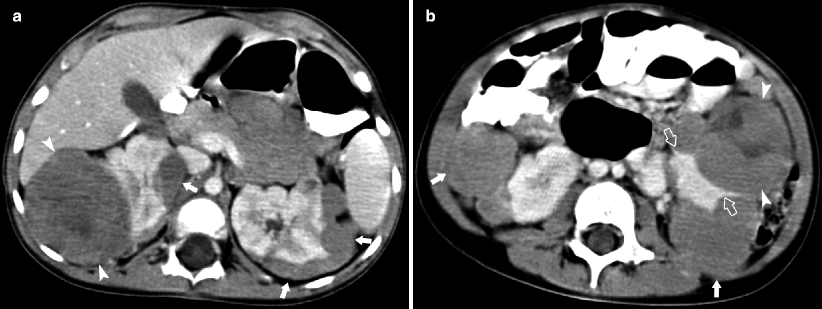
Fig. 10.2
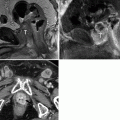
A 2-year and 10-month-old female presented with abdominal distention. (a, b) Contrast-enhanced CT (CECT) demonstrates multiple homogenously enhancing lenticular masses (closed arrows) in the peripheral cortex on both kidneys consistent with perilobar nephrogenic rests. Distortion of the central renal parenchyma of the left kidney resembling a staghorn (open arrow) as described by Rohrschneider et al. [7] is seen. Heterogeneous spherical masses seen in the upper pole of the right kidney and the lower pole of the left kidney (arrowheads) were proven to be favorable histology Wilms tumors in this child with nephroblastomatosis
Nephroblastomatosis refers to multifocal or diffuse distribution of nephrogenic rests [8]. Nephrogenic rests may be solitary or diffuse. Two subtypes exist: perilobar and intralobar. Perilobar nephrogenic rests occur in the peripheral renal cortex and are multiple whereas intralobar nephrogenic rests are usually solitary or few in number and occur anywhere within the renal lobe [9]. Wilms tumor is commonly associated with intralobar nephrogenic rests, although they occur less frequently than perilobar rests. Nephrogenic rests are associated with unilateral Wilms tumor in 41 % of cases and with bilateral Wilms tumor in 99 % of cases. Intralobar nephrogenic rests are associated with such Wilms tumor risk factors as Drash (ambiguous genitalia and progressive renal failure), sporadic aniridia, and WAGR [2, 9]. Perilobar rests are associated with Beckwith-Wiedemann syndrome and hemihypertrophy [9]. Nephrogenic rests exhibit homogenous appearance on ultrasound (US), and pre- and post-contrast computed tomography (CT) and magnetic resonance imaging (MRI) [7]. A heterogeneous or enlarging mass in a patient with nephroblastomatosis is concern for transformation to Wilms tumor.
Stage 1 and 2 Wilms Tumor
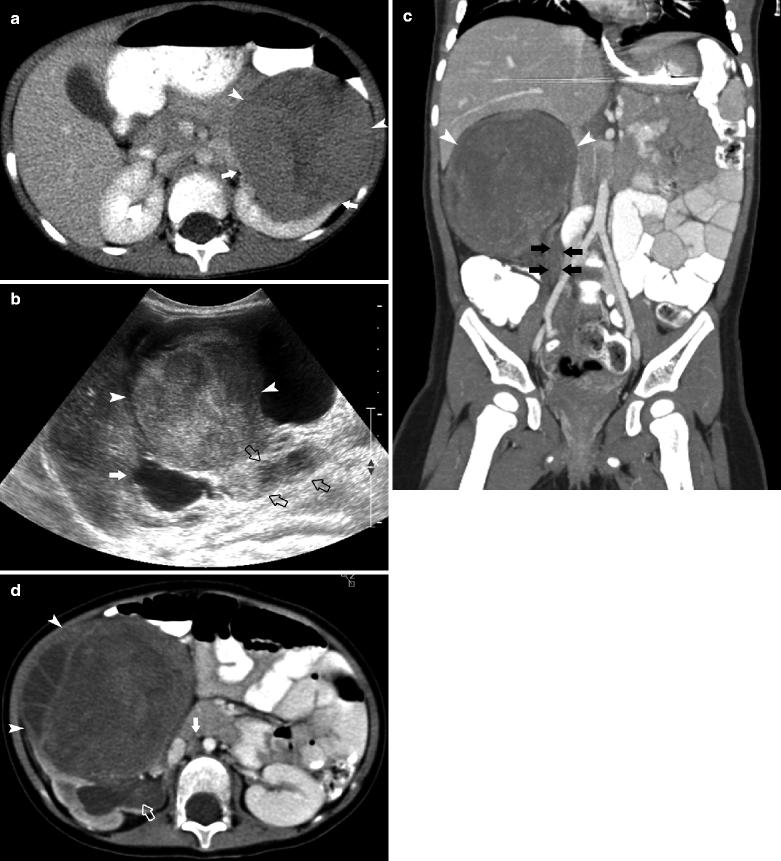
Fig. 10.3
A left renal mass was incidentally discovered on ultrasound evaluation of a 3.5-year-old male for suspected splenomegaly. Axial CECT image (a) demonstrates a heterogeneous mass in the upper half of the left kidney (arrowheads). A rim of normal renal parenchyma (claw sign; arrow) is present. No regional extension or distant metastases was seen. Surgery and pathological specimen confirmed stage 1 favorable histology Wilms tumor. A 2-year-old female with bloody stool and abdominal pain was noted to have a distended abdomen. Sagittal US (b) and coronal CT (c) demonstrate a heterogeneous mass originating in the right mid kidney (arrowhead) and a polypoid mass (arrows) in the proximal right ureter. Axial CT (d) demonstrates a complex mass with peripheral septations (arrowheads), interaortocaval lymph node (arrow), and obstruction of the right renal pelvis with layering debris (open arrow). Pathologic examination revealed tumor invasion into renal pelvis and proximal ureter without extension of tumor to adjacent lymph nodes. Chronic pyelonephritis from obstruction and reactive lymph nodes were found. The presence of ureteral extension did not affect stage 2 designation of the tumor. Ureteral extension of Wilms tumor is rare, but should be suspected in patients with gross hematuria, hydronephrosis, or nonfunctioning kidney. Gross hematuria, unlike microscopic hematuria, is uncommonly found at presentation of Wilms tumor [10]
Ruptured Wilms Tumor
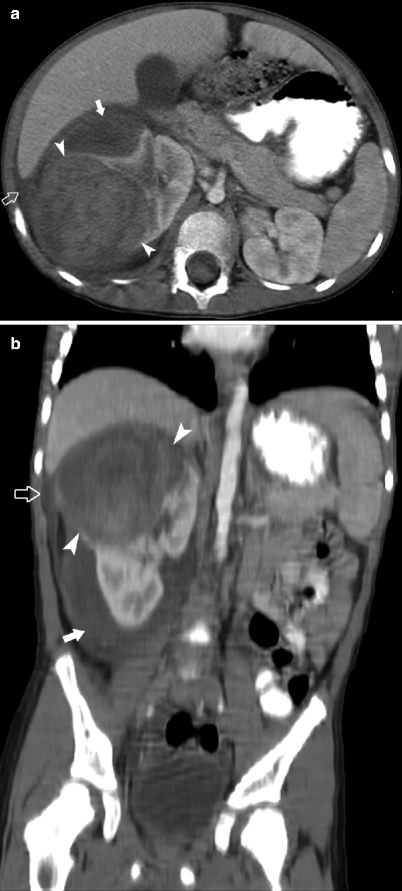
Fig. 10.4
A 3-year-old male presents with 2-week history of abdominal pain and 2-day history of marked abdominal swelling. Axial CT and coronal images (a, b) show a large heterogeneous mass (arrowhead) extending from the right upper pole. Subcapsular fluid (closed arrow) alters the renal contour. Subcapsular fluid and fluid within Gerota’s fascia represent the contained component of a rupture Wilms tumor. Fluid extending along the right paracolic gutter (open arrow) into the pelvis represents free spillage and contamination. Tumor spillage either before or during surgery classifies the tumor as stage III. In cases of contained retroperitoneal rupture at diagnosis there is an increased risk of peritoneal contamination at surgery. Localized flank radiation is administered in cases of contained retroperitoneal rupture. Total abdominal radiation is given in cases of peritoneal contamination [11]
Stage 4 Wilms Tumor
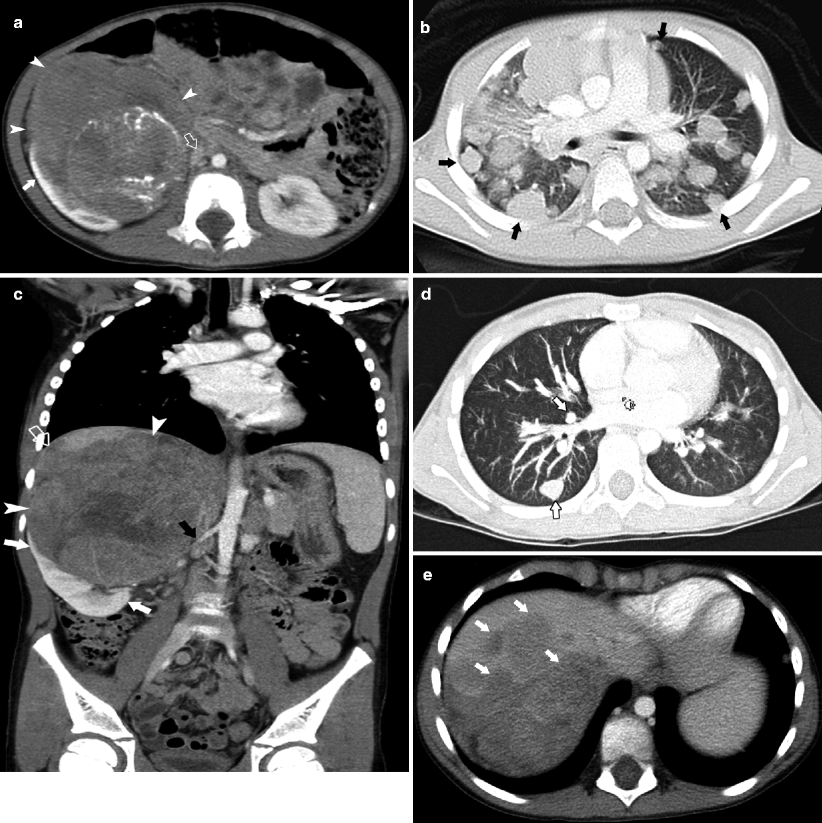
Fig. 10.5
A 2.5-year-old male presented with 2-day history of microscopic hematuria. Axial CT (a) demonstrates a large right renal mass with calcification (arrowheads). The IVC (open arrow) is compressed. Calcification is present in less than 10 % of Wilms tumor. Extensive pulmonary metastases are present (arrows) (b). No intravascular tumor thrombus is present. Surgery and pathology revealed stage IV, favorable histology Wilms tumor. A 7-year-old male presented with fever, fatigue, tachycardia, and paleness. Electrocardiography showed displacement of IVC. Coronal CT images (c) demonstrate a large right renal mass (arrowheads), with normal lower pole parenchyma creating a claw sign (white arrows), direct invasion of the liver and encasement of the right renal artery (black arrow). Unilateral renal involvement and distant spread of tumor to the lung and liver (arrows in d, e) classifies this as stage 4 disease. Hepatic metastases occur in 10–15 % of children with Wilms tumor [12]
Imaging studies and surgical and pathological findings at operative intervention determine the stage of Wilms tumor. Additional prognostic criteria are histology, favorable triphasic histology (blastema, stromal, and epithelial elements), or unfavorable histology (anaplastic sarcomatous elements). Pulmonary metastases are seen in 15–20 % of cases [12].
Bilateral Wilms Tumor
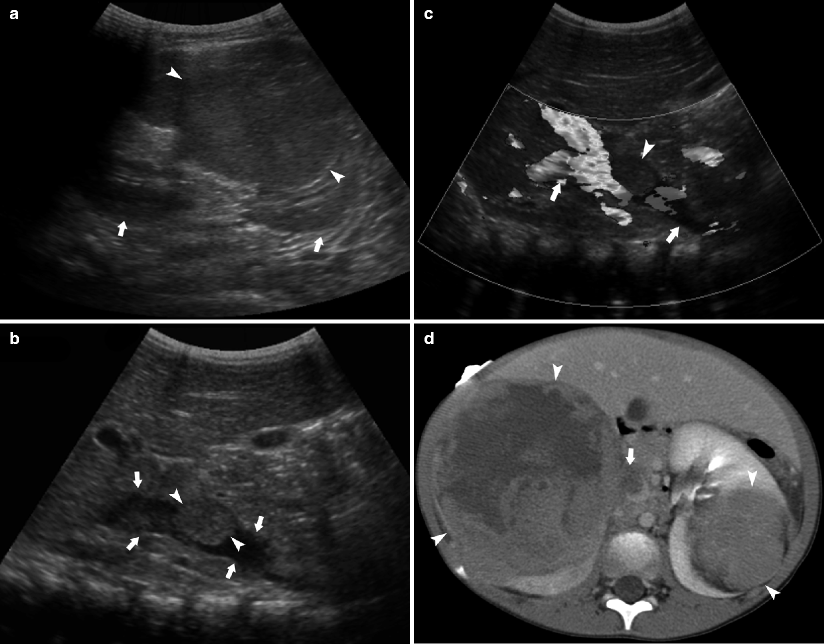
Fig. 10.6
A 3-year-old female presented with hematuria and abdominal pain. Sagittal ultrasound demonstrates mass (arrowhead) in the mid zone of the left kidney (arrows in a). A large right upper pole renal mass was also present. Evaluation of the inferior vena cava (arrow) reveals an echogenic tumor thrombus (arrowhead) at the level of the right renal vein Color Doppler imaging confirms the thrombus is nonocclusive (b, c). CECT (d) demonstrates heterogeneous right renal mass (arrowheads) and smaller left renal mass (arrowhead). Filling defect in the IVC (arrow) represents tumor localized tumor thrombus. The thrombus did not extend cephalad toward the right atrium
The presence of bilateral disease at diagnosis upstages patients to stage V [13]. In 5 % of patients, bilateral tumors are synchronous. Synchronous tumors occur more frequently in females and at a younger age (mean age, 2.5 y) [8]. Metachronous bilateral Wilms tumors are seen more frequently in children with initial presentation at less than 1 year of age and in patients with Beckwith-Wiedemann syndrome, hemihypertrophy, or congenital aniridia [8]. Bilateral disease represents a surgical challenge. Nephron-sparing surgery and partial nephrectomies or unilateral nephrectomy and contralateral nephrectomy are frequently performed.
Extrarenal Wilms Tumor
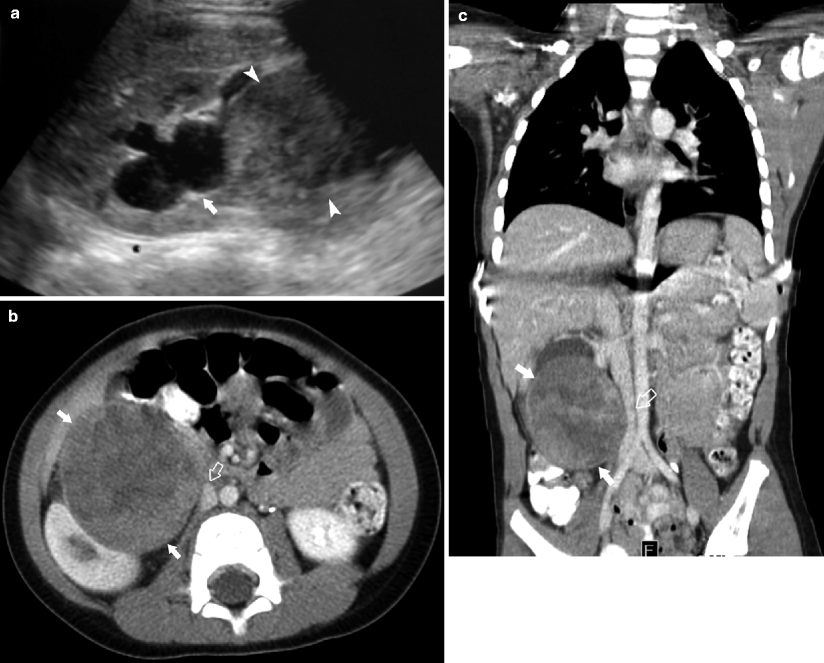
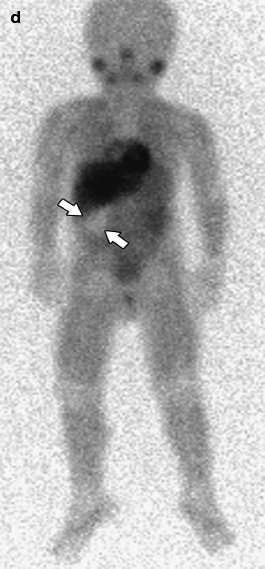
Fig. 10.7
A 3-year-old male was noted to have a right flank mass on physical examination. Sagittal ultrasound (a) demonstrates a solid mass (arrowheads) inseparable from the right kidney and causing obstruction at the level of the renal pelvis (arrow). Contrast-enhanced axial (b) and coronal reformatted CT (c) images demonstrate a heterogeneous mass extrinsic to the lower pole of the right kidney (arrows). The mass compresses the renal parenchyma and adjacent IVC (open arrow). No calcification is seen. MIBG scan (d) performed to evaluate preoperative diagnosis of paraspinal neuroblastoma demonstrates a photopenic area corresponding to the mass (arrows). Surgical exploration revealed a mass adjacent to the renal hilum. The renal capsule was intact and total gross resection was performed. An extrarenal Wilms tumor was diagnosed on pathology
Extrarenal Wilms tumor is an exceedingly rare tumor with approximately 200 cases reported in the literature involving pediatric and adult patients [14]. Retroperitoneal and pelvic locations are more common and may lead to incorrect diagnosis of neuroblastoma or ovarian lesion. Extrarenal Wilms tumor is believed to occur in any site that develops from pronephros, mesonephros, or metanephros [14].
Clear Cell Sarcoma of the Kidney
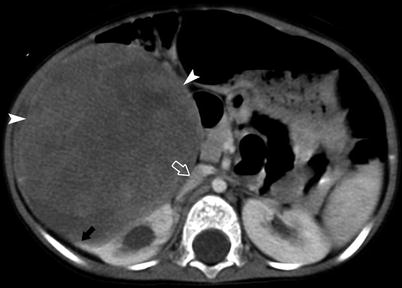
Fig. 10.8
An abdominal mass was incidentally discovered in a 19-month-old male. A CECT demonstrates a large right mass (arrowheads) demonstrating a claw sign (black arrow) indicating renal origin of the mass. The IVC is compressed (open arrow). No tumor thrombus is identified in the renal vein or IVC. No local adenopathy is present. No pulmonary, hepatic, and lymph node metastases were present. The radionuclide bone scan in this patient was negative. A definitive resection, radical right nephrectomy followed by adjuvant chemotherapy, was the treatment course. Pathology revealed clear cell sarcoma of the kidney (CCSK)
CCSK is an aggressive renal tumor, pathologically distinct from Wilms tumor. On imaging, however, CCSK cannot be differentiated from Wilms tumor. The peak incidence is 1–4 years old with a male predominance [2]. CCSK is known as the “bone metastasizing renal tumor of childhood” [12]. 99mTc-MDP bone scintigraphy is essential for staging this disease. In addition to skeletal metastases that occur in 40–60 % of patients [12], CCSK spreads to lymph nodes, brain, liver, and lung [2]. Patients with CCSK have a longer surveillance period than those diagnosed with Wilms tumor. The prognosis is worse with a long-term survival of 60–70 % [13].
Rhabdoid Tumor
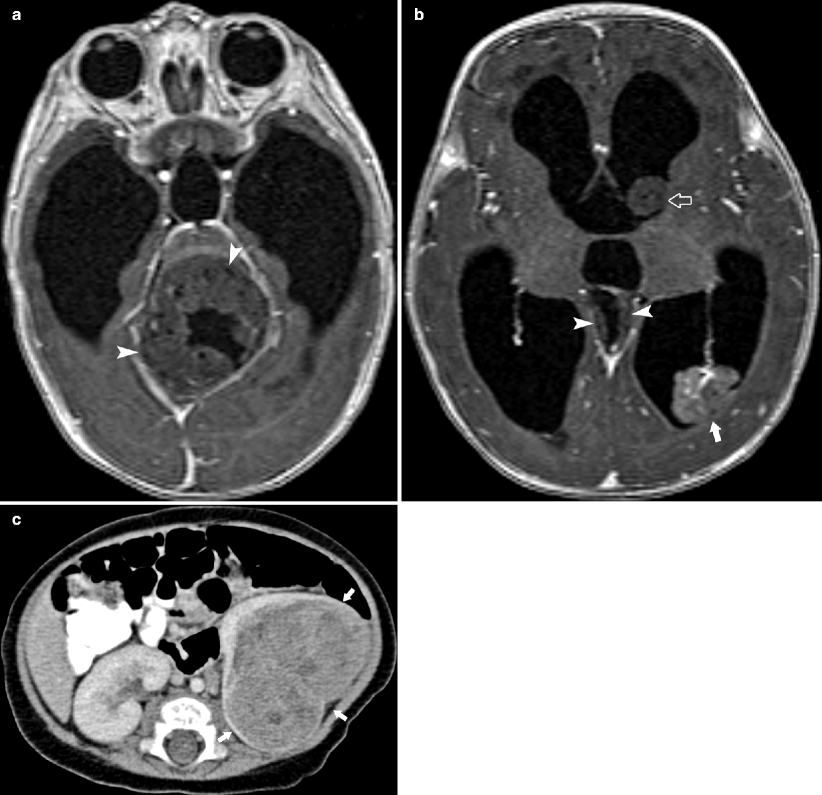
Fig. 10.9
A 5-month-old female presented with intermittent opisthotonic posturing and rapidly increasing head circumference. Axial post-contrast MR demonstrates mass originating in the cerebellar vermis resulting in obstructive hydrocephalus of the third and lateral ventricles (arrowhead in a, b). A separate multilobular-enhancing mass in the posterior atrium of the left lateral ventricle (open arrow) is contiguous with the choroid plexus and a third nonenhancing mass is seen along the ependymal surface of the left lateral ventricle (arrow in b). Resection of posterior fossa mass revealed an atypical teratoid rhabdoid tumor. Abdominal CT (c) performed for extent of disease demonstrated a large heterogeneously enhancing mass in the left kidney involving the renal hilum (arrows). Left nephrectomy revealed an rhabdoid kidney of the tumor (RTK)
RTK are extremely aggressive neoplasms [2, 12]. More the 50 % of patients with rhabdoid tumor of the kidney are younger than 1 year old [2, 12]. The 18-month survival rate is only 20 % [2]. RTK are associated with primary synchronous or metachronous, midline, or posterior fossa tumors in 10–15 % of cases [12]. Metastases to the lung are most common, followed by liver, lymph nodes, bone, and brain [2, 12]. On imaging, RTK are large heterogeneous masses arising in the renal hilum. Subcapsular fluid collections representing necrosis and/or hemorrhage and linear calcifications outlining the tumor lobules may be present [2, 12].
Medullary Renal Cell Carcinoma
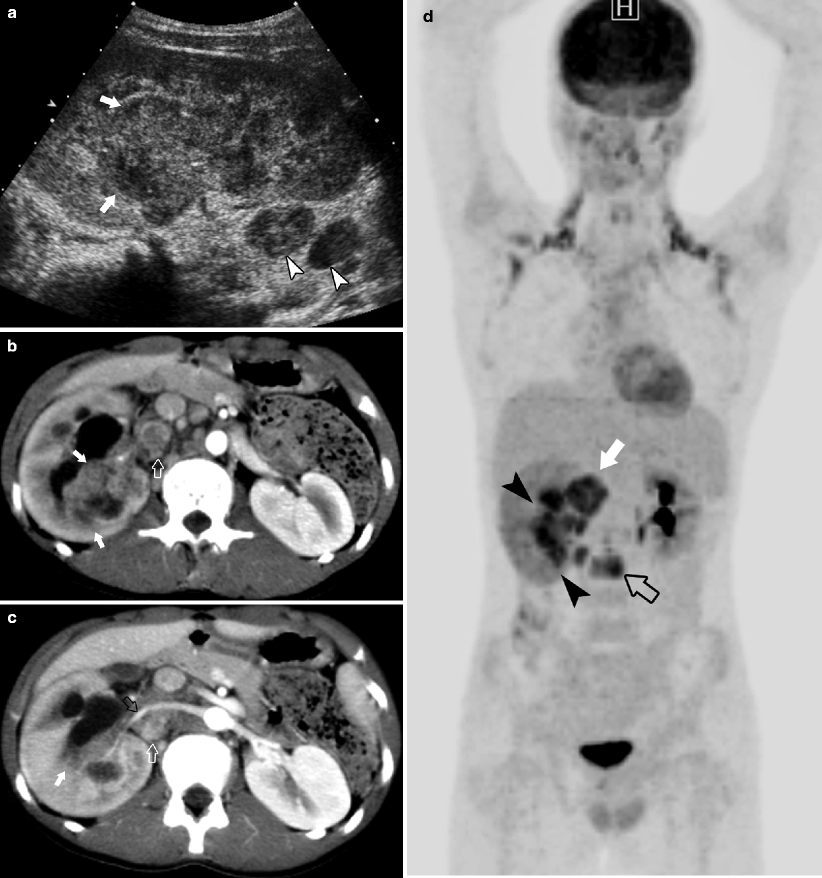
Fig. 10.10
A 13-year-old male with previously undiagnosed sickle cell trait presented with a 2-month history of intermittent hematuria, weight loss, and back pain. Ultrasound (a) performed for evaluation of hematuria demonstrates solid mass (arrows) involving the right renal pelvis and abnormal retroperitoneal lymph nodes (arrowheads). CECT (b) demonstrates a heterogeneous mass in the renal medulla (arrows). The mass (arrow) extends into the renal pelvis, obstructing the collecting system. Right paraaortic adenopathy (white open arrow) displaces the renal artery (black open arrow) (c). Maximum intensity projection (MIP) image from FDG-PET (d) demonstrates FDG avid infiltrative tumor in the right renal pelvis (arrowheads), adenopathy adjacent to the renal hilum (arrow), and metastases to L2 (open arrow). Pathology confirmed diagnosis of medullary renal cell carcinoma (RMC)
RMC occurs in patients with sickle cell trait or hemoglobin sickle cell (SC) disease, not homozygous sickle cell (hemoglobin SS) disease [12, 15]. Painless gross hematuria is the most common presentation in patients who range in age from 10 to 40 years old (mean, 22 y) [15]. The enlarged kidney characteristically maintains its reniform shape. The tumor replaces the renal pelvis infiltrating the renal sinus and causes caliectasis. RMC occurs more frequently on the right side [15]. Hemorrhage and necrosis are consistent findings. Metastatic spread is to regional lymph nodes, lung, adrenal gland, and liver [15, 16]. RMC is an aggressive tumor with poor response to chemotherapy or radiation. The average survival from time of diagnosis is 15 weeks [2].
Renal Cell Carcinoma
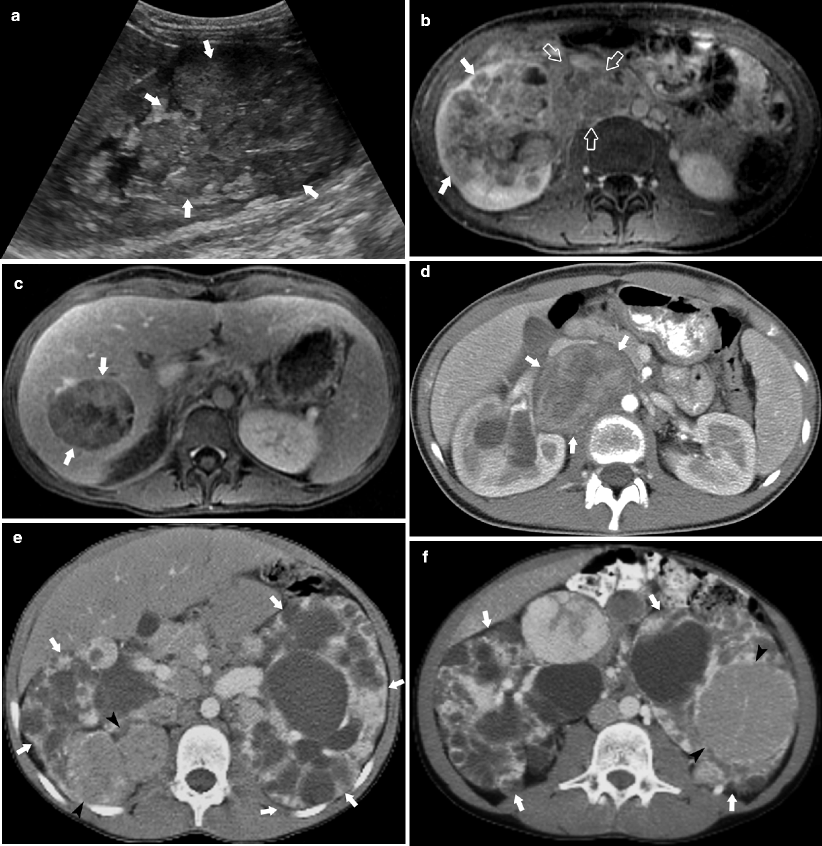
Fig. 10.11
A 14-year-old female presented with a 6-month history of anemia and intermittent microscopic hematuria. Sagittal ultrasound (a) demonstrates an infiltrative right lower pole renal mass (arrows). Post-contrast axial T1-weighted MR (b, c) demonstrates the right lower pole renal mass (arrows), spread to adjacent lymph nodes (open arrows) and metastases to the right lobe of the liver (arrows). Axial CECT (d) demonstrates retroperitoneal lymph nodes (arrows) displacing the right renal vein and IVC. Pathology revealed XpII/TFE 3 translocation-associated RCC, a recently described entity that accounts for almost one third of pediatric RCC [17]. An 11-year-old male with known tuberous sclerosis and enlarged cystic kidneys (arrows) (e, f) and enlarging renal masses (arrowheads) on surveillance CT. Biopsy of the enlarging renal masses confirmed the suspected diagnosis of renal cell carcinoma (RCC)
RCC represents less than 7 % of all malignant renal tumors in children usually occurring within the first two decades of life [12]. RCC in children is associated with tuberous sclerosis, von Hippel Lindau syndrome, urogenital malformations, and treated neuroblastoma. The mean interval between treatment of neuroblastoma and the diagnosis of RCC is 11 years [18]. Metastases to liver, lung, or brain are reported in 20 % of patients at the time of diagnosis [2]. Imaging alone is insufficient to differentiate RCC from Wilms tumor. Although the incidence of Wilms tumor compared to RCC is nearly 30:1 in childhood, the incidence is nearly equal in the second decade of life [2]. Calcification is present in 25 % of cases of RCC compared to 9 % of Wilms tumor [2].
Leukemia
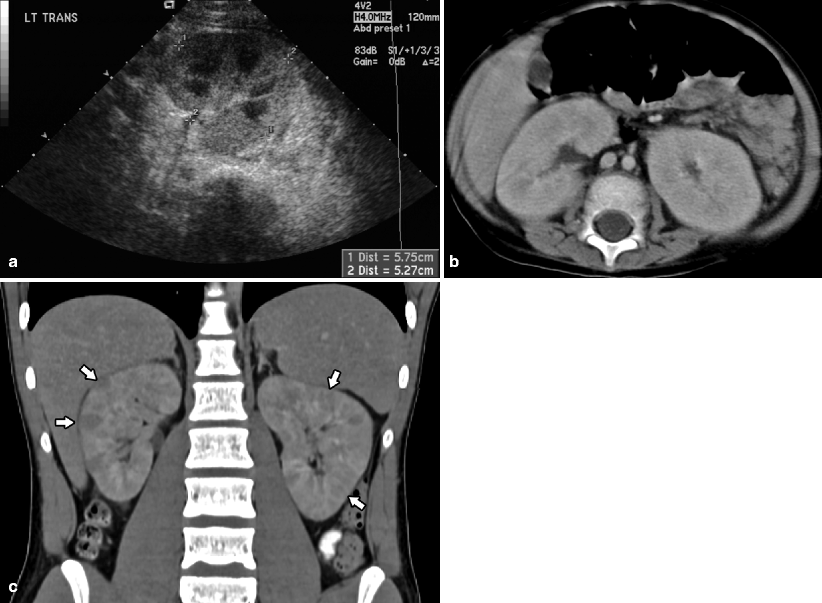
Fig. 10.12
Ultrasound (a) and CT (b) performed in a newly diagnosed 2-year-old male with ALL demonstrates homogenously enlarged kidneys that are greater than 95 % of the predicted size limits for the patient’s age. Imaging of renal involvement in leukemia may demonstrate bilaterally enlarged kidneys with loss of corticomedullary differentiation and masses, solitary or multiple [12]. The kidneys are a sanctuary site for leukemia and imaging findings may occur during times of active disease as well as in bone marrow remission. A 17-year-old presented with a 1-month history of cervical adenopathy and fatigue. Coronal reconstruction CT (c) performed prior to the pathologic confirmation of suspected diagnosis of T cell lymphoblastic leukemia demonstrates bilaterally enlarged kidneys with multiple low attenuation infiltrative lesions (arrows)
Lymphoma
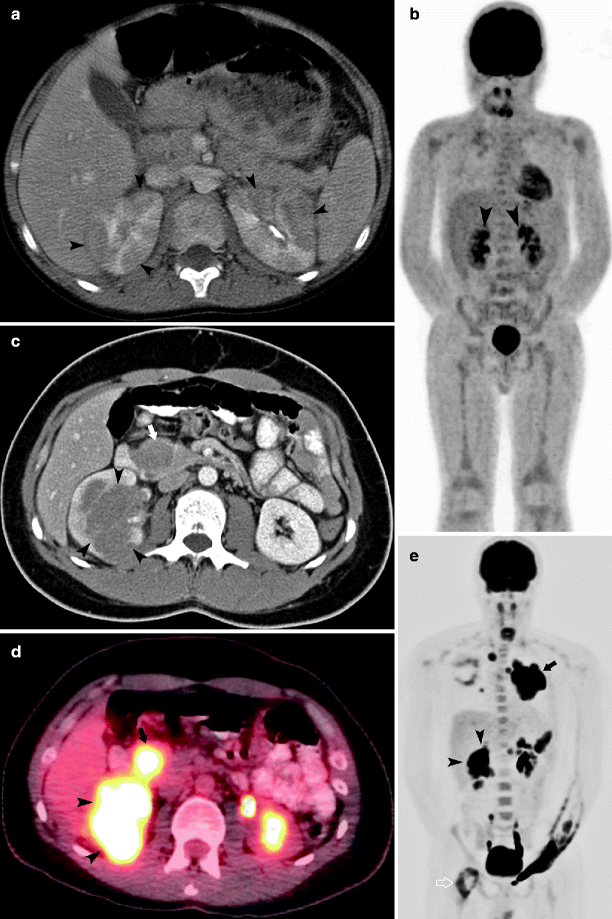
Fig. 10.13
Axial CT (a) performed in a 7-year-old male diagnosed with Burkitt’s lymphoma demonstrates diffuse low attenuation lesions (arrowhead) relative to normal renal parenchyma that distort the contour of the kidneys. MIP image from the FDG-PET scan (b) demonstrates FDG avidity in the renal lesions (arrowheads). In addition to diffuse lesions, lymphoma can present as a solitary renal mass. Axial CT (c) in a 17-year-old male diagnosed with diffuse large B-cell lymphoma demonstrates a large right renal mass (arrowheads). Retroperitoneal adenopathy (arrow) is apparent. The renal mass (arrowheads) and adjacent adenopathy are highly FDG avid as seen on an axial fused PET-CT image (d). MIP image (e) demonstrates renal involvement (arrowheads), large anterior mediastinal mass (arrow), and involvement of right proximal femur (open arrow)
Because the kidneys do not contain lymphatic tissue, renal involvement in patients with lymphoma is either by hematogenous spread or direct extension from retroperitoneal disease [12]. Renal involvement in lymphoma more commonly presents as multiple masses, with a solitary mass and homogenous nephromegaly less common. On MR, the lesions are homogenous, hypointense on T1-weighted image, and hyperintense on T2-weighted image with variable contrast enhancement [12]. Renal involvement is uncommon in Hodgkin lymphoma. Non-Hodgkin lymphoma, especially Burkitt and T-cell lymphoblastic lymphoma, frequently involves the kidney [19].
Rhabdomyosarcoma of the Bladder/Prostate
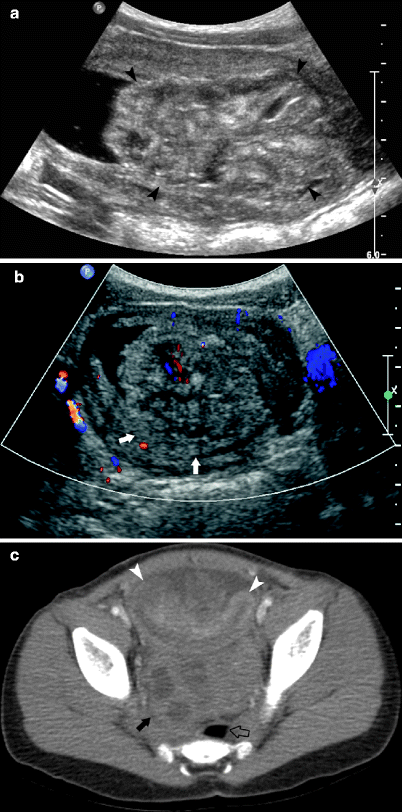
Fig. 10.14
A 6-year-old male presented with hematuria and dysuria. Sagittal ultrasound of the bladder demonstrates a large heterogeneous mass (arrowheads) arising from the base of the bladder (a). Transverse color Doppler ultrasound image reveals minimal flow within the mass that is inseparable from the bladder wall (arrow) (b). Pathologic diagnosis was embryonal rhabdomyosarcoma (RMS), botryoid subtype. A 3-year-old male presented with abdominal pain, distention, and hematuria (c). Axial image from CECT demonstrates a heterogeneously enhancing grapelike intravesical mass (arrowheads). A retrovesical mass (closed arrows) displaces the rectum (open arrow). As is often the case in patients with large RMS, the origin of the lesion, bladder versus prostate RMS, is not possible to determine. Biopsy confirmed diagnosis of embryonal RMS, botryoid subtype
RMS of the bladder and prostate represent 10 % of all cases of RMS usually occurring in children younger than 9 years old. In cases of only bladder involvement, the boy–girl ratio is 3:1 [20]. Staging of RMS is extremely complex [21, 22]. Site and histology are important factors. Nonbladder/nonprostate genitourinary primaries have a favorable prognosis, unlike primary bladder and prostate RMS. Dysuria, hematuria, and urinary retention are all presenting symptoms. Bladder/prostate RMS may be infiltrative, polypoid, or resemble a cluster of grapes (sarcoma botryoides). Ninety percent of bladder RMS is of the embryonal subset. Botryoid subtype of embryonal RMS are associated with a more favorable prognosis [21, 22]. Treatment with chemotherapy and radiation therapy to reduce tumor bulk permits bladder function preservation surgery without sacrificing cure rates [21, 23].
Neuroblastoma
Neuroblastoma (NB) is the most common extracranial neoplasm in childhood and represents 8 % of all pediatric malignancies [24, 25]. It is the most common tumor of infants [25]. Although most tumors occur in children from 2 to 5 years old, NB may also be seen in newborns, adolescents, and adults [26]. NB derives from neural crest cells that give rise to the sympathetic nervous system. Therefore, NB can occur anywhere along the sympathetic chain or in the adrenal medulla. The majority of tumors are found in the adrenal medulla or other retroperitoneal sites [27]. However, primary tumors may occur in the neck (5 %) [28], posterior mediastinum (20 %), and pelvis (5 %).
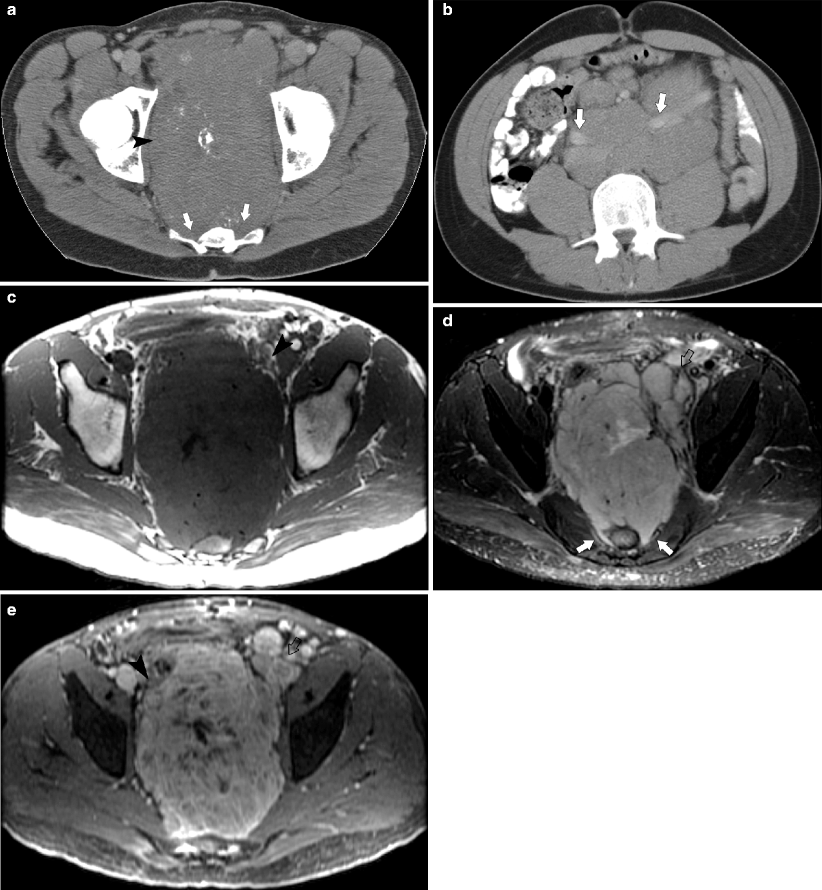
Fig. 10.15
Pelvic neuroblastoma. (a, b) CT demonstrates a large partially calcified pelvic mass (arrowhead) extending into sacral foramina (arrows). Extensive accompanying retroperitoneal adenopathy, anteriorly displacing both common iliac arteries (arrows). (c–e) Axial MR. (c) T1-weighted image shows low signal mass with small areas of fluid or necrosis. Mass extends into sacral foramina. (d) T2-weighted fat saturation image shows mass with areas of fluid or necrosis (arrows) and nodes (open arrows). (e) T1-weighted post-contrast shows heterogeneous enhancement of mass (arrowhead) and nodes (open arrow) [27]
At least 60 % of patients have metastatic disease at diagnosis, most commonly in the following sites: bone, bone marrow, lymph nodes [29] (see Fig. 10.16), and liver [27]. Other less common metastatic sites include pleura (see Fig. 10.17), pulmonary parenchyma [30, 31], and the central nervous system (CNS) [32–36] (see Fig. 10.18).
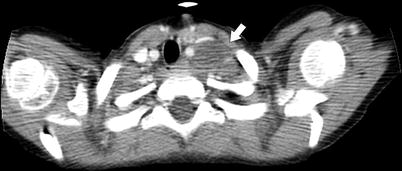
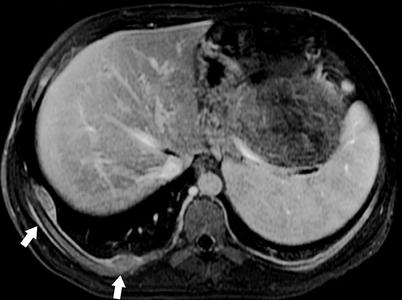
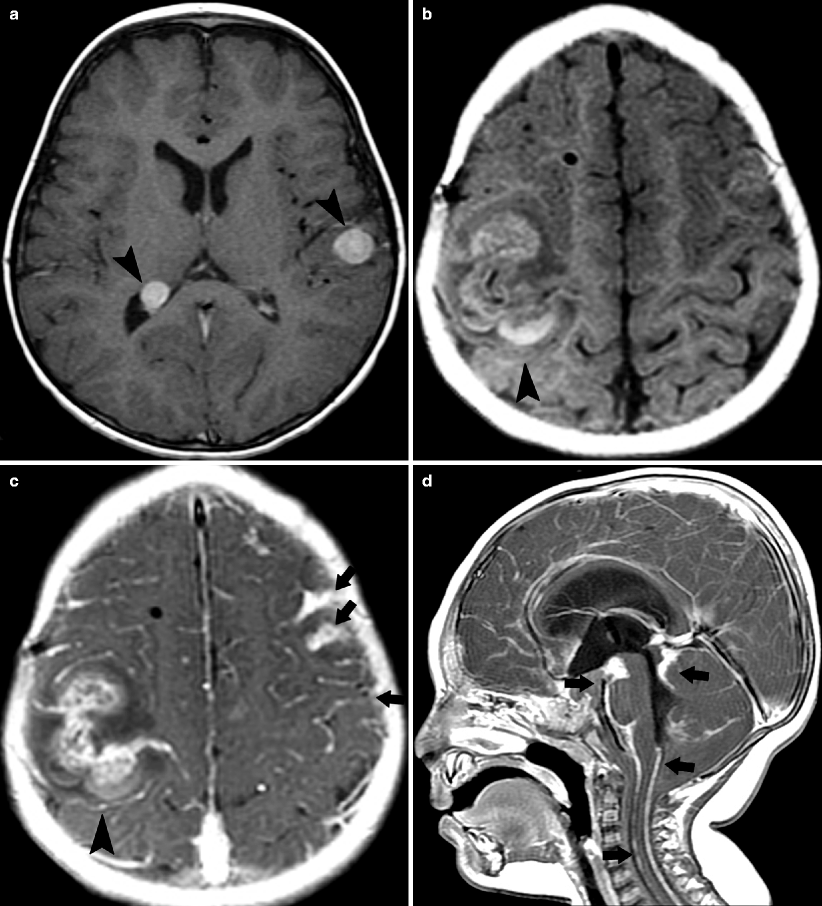
Fig. 10.16
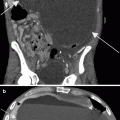
Left supraclavicular node. NB has a propensity for involving the left supraclavicular node (“Virchow” node; arrow). Anterior mediastinal and hilar adenopathy is uncommon. Sampling of this node to obtain tissue for diagnosis and evaluation of biologic markers precludes the need for abdominal surgery for diagnosis in patients with large unresectable abdominal masses [29]
Fig. 10.17
Pleural metastases. MR axial T-weighted post-contrast image demonstrates right pleural metastases (arrows). Pleural metastases are uncommon in neuroblastoma, usually occurring at relapse [27]. This 10-year-old male presented with these findings
Fig. 10.18
CNS metastases. MR. (a) Axial T1-weighted post-contrast image demonstrates left intracerebral and right intraventricular metastases (arrowheads). (b) Second patient with axial T1-weighted pre-contrast scan showing large right hemorrhagic metastasis that enhances on post-contrast image. (c) Note also the leptomeningeal disease (arrows). (d) Sagittal T1-weighted post-contrast image from a third patient shows hydrocephalus and extensive leptomeningeal disease (arrows). CNS metastases are uncommon and usually occur at relapse [32, 33, 36]
Staging and Prognosis
The International Neuroblastoma Staging System (INSS) was devised as a staging system, taking into account radiological findings, surgical resectability, and metastatic disease [37].
1 | Localized tumor, complete gross resection, negative nodes bilaterally at surgery |
2A | Localized tumor, incomplete gross resection, all nodes negative |
2B | Localized tumor, with or without complete gross resection, positive ipsilateral nodes, negative contralateral nodes |
3 | Tumor across midline, with or without regional node involvement; unilateral tumor with contralateral nodes; or midline tumor with bilateral nodes |
4 | Distant metastases to lymph nodes, bone, bone marrow, liver, or other organs |
4S | <1 year old, localized tumor (1, 2a, or 2b), spread limited to skin, liver, or bone marrow (<10 %) |
However, other factors also affect prognosis in neuroblastoma, including age at diagnosis, site of primary, histology, and certain biologic factors that have been found to be linked to prognosis [27]. Patients younger than 1 year old at diagnosis have a significantly increased survival. Also, extraabdominal site of primary and favorable Shimada histology [38] also are associated with improved survival. Several biologic factors have been identified that are associated with prognosis [39]. Amplification (> 10 copies) of MYCN oncogene, a segment of DNA on chromosome 2, closely correlates with poor prognosis. Most patients younger than 1 year old do not have amplification [40]. DNA ploidy in infants also relates to prognosis [41]. DNA index greater than 1 in children younger than 1–2 years old is associated with a good prognosis, whereas DNA ploidy equal to or less than one is linked to poorer survival [40]. The set of markers in a given patient does not change with time so that favorable forms do not convert into unfavorable disease or vice versa [42]. Combining the INSS with these other important factors allows for determining low, intermediate, and high-risk disease. Therapy and prognosis are based on this combined risk classification system [40, 43–45].
Low risk | Intermediate risk | High risk |
|---|---|---|
Stage I (completely removed) | Stage II no MYCN but <50 % removed | Stage II, III, IV, or IVS with MYCN |
Stage II No MYCN and >50 % resection | Stage III <18 months, no MYCN | Stage III >18 months and unfavorable histology |
Stage IVS no MYCN, DNA index >1, favorable histology and no symptoms | Stage III >18 months with no MYCN and favorable histology Stage IV <12 months and no MYCN | Stage IV 12–18 months, no MYCN unfavorable histology and DNA index 1 |
Stage IV 12–18 months, no MYCN, favorable histology, and DNA index >1 | Stage IV >18 months | |
Stage IVS no MYCN, with either unfavorable histology or DNA index of 1 | ||
95 % Survival | >90 % Survival | 30–40 % Survival |
Clinical Presentation
Children with neuroblastoma present with a wide variety of symptoms, frequently associated with metastatic disease. Because the majority of patients have metastatic disease at presentation, many of them present with pallor, weight loss, and irritability. A prominent mass in the abdomen may be related to an enlarged liver from metastatic deposits or from the large primary tumor. The tumor may extend through neural foramina into the extradural space (dumbbell tumor) causing neurologic symptoms from cord compression. Involvement of the sphenoid bone may cause ecchymoses around the orbit from hemorrhage into the surrounding soft tissues and may be accompanied by orbital proptosis (“raccoon eye”) [42, 46].
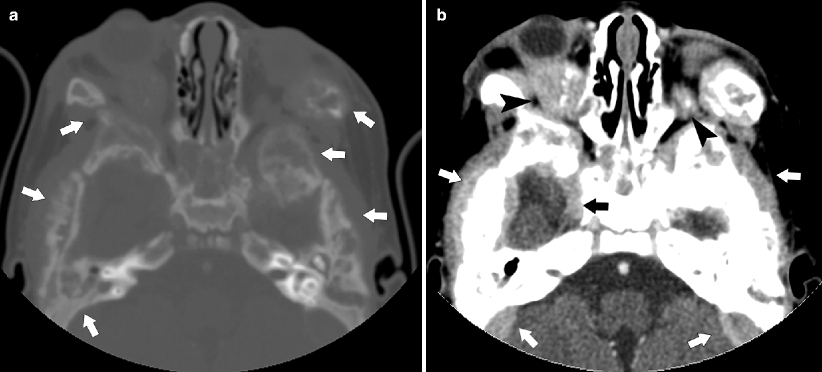
Fig. 10.19
Extensive bone and soft tissue metastases. This 2-year-old boy presented with proptosis, “raccoon eyes,” and blindness due to extensive bone and accompanying soft tissue disease that extended into the orbit and compressed the optic nerves. (a) CT bone window. Virtually all the bones of the calvarium are involved (arrows). (b) Soft tissue window at same level shows marked soft tissue involvement (arrows) from the underlying bone disease, extending into the orbits and causing right proptosis (arrowheads) [42, 46]
Patients with advanced disease at diagnosis frequently present with diffuse bone pain, refusal to bear weight, and limping from bony metastases. Cervical and superior sulcus tumors may present with Horner’s syndrome 5. Rarely, compression of both renal arteries by tumor and nodes may lead to hypertension [46].
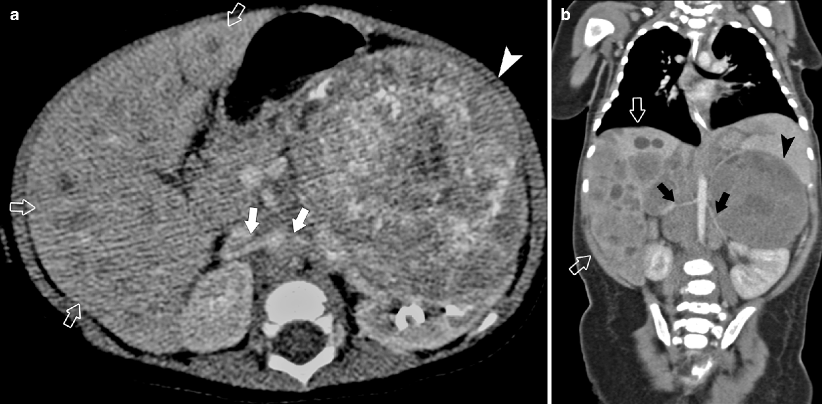
Fig. 10.20
A 2-year-old child with hypertension due to nodal encasement of both renal arteries. CT. Axial (a) and coronal (b) images show large partially calcified left adrenal mass (arrowheads) and nodes, inferiorly displacing the left kidney and also encasing the aorta, superior mesenteric arteries, and both renal arteries (arrows). Multiple hepatic metastases are also present (open arrow). Hypertension is uncommon in NB and, when present, is secondary to compression of the renal arteries, not from catecholamine secretion. When the nodes decrease in size, the hypertension abates [46]
Intractable diarrhea may be the presenting symptom in children with tumors that secrete vasoactive intestinal peptide (VIP). Diarrhea abates after tumor resection [47]. Some patients with myoclonic encephalopathy of infancy (dancing feet, dancing eyes) may have underlying neuroblastoma [48, 49]. Neuroblastoma may also be identified on routine prenatal ultrasonographic screening [50, 51] or by urine catecholamine screening [52–55].
IVS NB
Patients with stage IVS disease are a special group of patients with a survival rate of greater than 90 % [40, 44




Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
