CHAPTER EDITOR
DONALD P. FRUSH
HISTORY
An 8-year-old boy with pleuritic chest pain and 39°C temperature.
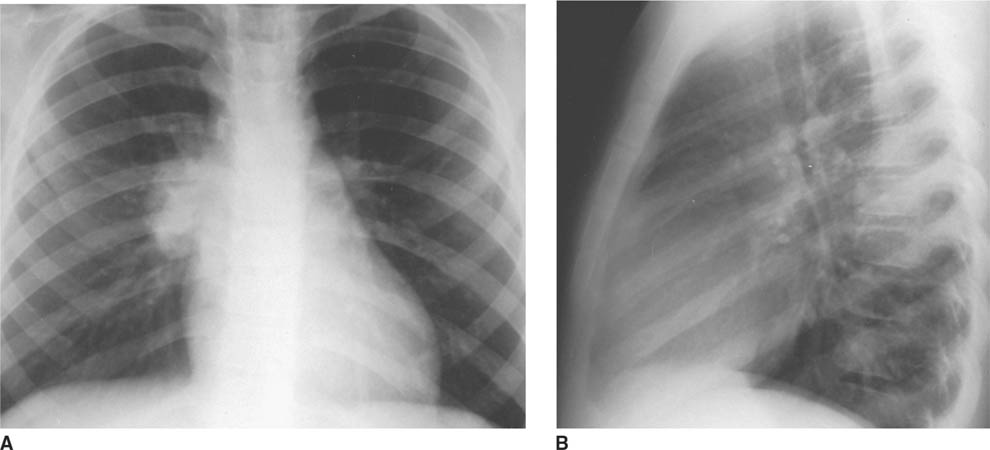
 FIGURES 8-1A and 8-1B Frontal (A) and lateral (B) chest radiographs. A well-defined mass is present in the right lung. Notably, the right hilum is not obscured (i.e., the hilar overly sign is present) and thus the mass is not hilar in location. This mass is posterior on the lateral view, projected over the upper mid-thoracic spine and is in the superior segment of the right lower lobe.
FIGURES 8-1A and 8-1B Frontal (A) and lateral (B) chest radiographs. A well-defined mass is present in the right lung. Notably, the right hilum is not obscured (i.e., the hilar overly sign is present) and thus the mass is not hilar in location. This mass is posterior on the lateral view, projected over the upper mid-thoracic spine and is in the superior segment of the right lower lobe.
 Hilar adenopathy: This diagnosis is incorrect because the right hilum is seen as separate from the “mass.” Therefore, the mass must be anterior or posterior in the right hemithorax.
Hilar adenopathy: This diagnosis is incorrect because the right hilum is seen as separate from the “mass.” Therefore, the mass must be anterior or posterior in the right hemithorax.
 Plasma cell granuloma (postinflammatory or inflammatory pseudotumor; (fibrous) histiocytoma, myo-fibroblastic tumor, xanthogranuloma): Plasma cell granuloma can present as a mass, with calcification in up to 25% of cases. Children are usually asymptomatic or have minimal symptoms. This diagnosis is possible in the case presented and cannot be excluded on the basis of the radiographs, but it is a relatively rare entity and so would be a lesser consideration than a more common entity that is compatible with the imaging findings.
Plasma cell granuloma (postinflammatory or inflammatory pseudotumor; (fibrous) histiocytoma, myo-fibroblastic tumor, xanthogranuloma): Plasma cell granuloma can present as a mass, with calcification in up to 25% of cases. Children are usually asymptomatic or have minimal symptoms. This diagnosis is possible in the case presented and cannot be excluded on the basis of the radiographs, but it is a relatively rare entity and so would be a lesser consideration than a more common entity that is compatible with the imaging findings.
 Bronchopulmonary foregut malformation: Such malformations—for example, parenchymal bronchogenic cyst or a pulmonary sequestration—are reasonable considerations on the basis of the findings shown. Cystic adenomatoid malformation is another malformation to be considered, but unlike in infants and young children, it rarely presents in older children or adults with air or air-fluid-filled cysts.
Bronchopulmonary foregut malformation: Such malformations—for example, parenchymal bronchogenic cyst or a pulmonary sequestration—are reasonable considerations on the basis of the findings shown. Cystic adenomatoid malformation is another malformation to be considered, but unlike in infants and young children, it rarely presents in older children or adults with air or air-fluid-filled cysts.
 Primary pulmonary malignancy: Although sarcomas and pulmonary (or pleuropulmonary) blastomas are the most likely primary tumors to produce these findings, these lesions are exceedingly rare and the diagnosis is unlikely.
Primary pulmonary malignancy: Although sarcomas and pulmonary (or pleuropulmonary) blastomas are the most likely primary tumors to produce these findings, these lesions are exceedingly rare and the diagnosis is unlikely.
 Metastasis: This diagnosis is an unlikely one because there is no history of malignancy. Furthermore, a single, large metastasis would be unusual.
Metastasis: This diagnosis is an unlikely one because there is no history of malignancy. Furthermore, a single, large metastasis would be unusual.
 Round pneumonia: Round pneumonia is a very common cause of a pulmonary mass in children. This entity is much more common than a bronchopulmonary fore-gut malformation. Round pneumonia would be readily distinguishable from a cystic congenital lung abnormality based on central low density. However, it would be indistinguishable by imaging features alone from a solid congenital lung abnormality. Nonetheless, on the basis of probability alone, round pneumonia is much more likely than a solid congenital lung abnormality.
Round pneumonia: Round pneumonia is a very common cause of a pulmonary mass in children. This entity is much more common than a bronchopulmonary fore-gut malformation. Round pneumonia would be readily distinguishable from a cystic congenital lung abnormality based on central low density. However, it would be indistinguishable by imaging features alone from a solid congenital lung abnormality. Nonetheless, on the basis of probability alone, round pneumonia is much more likely than a solid congenital lung abnormality.
 Contusion: This diagnosis is not a reasonable consideration because there is no history of trauma.
Contusion: This diagnosis is not a reasonable consideration because there is no history of trauma.
 Round atelectasis: Round atelectasis is a consideration based on the round shape of the opacity but is usually basilar in location, whereas the lesion shown in this case is in the superior segment of the right lower lobe. Furthermore, round atelectasis is very rare in children, making this diagnosis unlikely.
Round atelectasis: Round atelectasis is a consideration based on the round shape of the opacity but is usually basilar in location, whereas the lesion shown in this case is in the superior segment of the right lower lobe. Furthermore, round atelectasis is very rare in children, making this diagnosis unlikely.
 Pleural effusion caused by “pseudotumor”: This entity is unlikely because it is usually sharply defined along at least one border in at least one projection, whereas the mass in the case shown here does not have sharp margins, making this diagnosis unlikely.
Pleural effusion caused by “pseudotumor”: This entity is unlikely because it is usually sharply defined along at least one border in at least one projection, whereas the mass in the case shown here does not have sharp margins, making this diagnosis unlikely.
 Hamartoma: This diagnosis is a reasonable consideration based on imaging findings alone but is usually asymptomatic and is relatively rare.
Hamartoma: This diagnosis is a reasonable consideration based on imaging findings alone but is usually asymptomatic and is relatively rare.
DIAGNOSIS
Round pneumonia in the superior segment of the right lower lobe
KEY FACTS
Clinical
 Almost all pulmonary masses in children are inflammatory and primarily due to infection.
Almost all pulmonary masses in children are inflammatory and primarily due to infection.
 Round pneumonia is a common cause of a pulmonary mass in children.
Round pneumonia is a common cause of a pulmonary mass in children.
 The mean patient age is 5 years. Round pneumonia is rare over the age of 8.
The mean patient age is 5 years. Round pneumonia is rare over the age of 8.
 The male:female gender ratio is 1:1.
The male:female gender ratio is 1:1.
 Streptococcus pneumoniae (pneumococcus) is the most common organism responsible for round pneumonia.
Streptococcus pneumoniae (pneumococcus) is the most common organism responsible for round pneumonia.
 Symptoms can be mild or nonspecific early in the clinical course.
Symptoms can be mild or nonspecific early in the clinical course.
 With a clinical history typical of pneumonia and recognition of a mass on imaging studies as round pneumonia, antibiotic treatment can be started without further imaging.
With a clinical history typical of pneumonia and recognition of a mass on imaging studies as round pneumonia, antibiotic treatment can be started without further imaging.
 In cases in which the clinical information is unclear, a pulmonary mass can be followed with radiography after therapy (6 to 8 weeks), unless symptoms persist or worsen.
In cases in which the clinical information is unclear, a pulmonary mass can be followed with radiography after therapy (6 to 8 weeks), unless symptoms persist or worsen.
Radiologic
 Round pneumonia is typically a well-defined, solitary (98% of cases), mass-like opacity.
Round pneumonia is typically a well-defined, solitary (98% of cases), mass-like opacity.
 85% are posterior; 65% of all round pneumonias are in the lower lobe.
85% are posterior; 65% of all round pneumonias are in the lower lobe.
 Two-thirds of the masses have sharply defined borders; one-third poorly defined borders. Mean age of the latter group is about 1 year older than in the former group.
Two-thirds of the masses have sharply defined borders; one-third poorly defined borders. Mean age of the latter group is about 1 year older than in the former group.
 Of those cases that are followed clinically and by imaging, 95% slowly resolve and 5% develop lobar pneumonia.
Of those cases that are followed clinically and by imaging, 95% slowly resolve and 5% develop lobar pneumonia.
 Air bronchograms are initially present in a minority of cases.
Air bronchograms are initially present in a minority of cases.
 Calcification is not a feature of this entity.
Calcification is not a feature of this entity.
 Unlike pneumonia in adults, pneumonia is often initially confined due to immature collateral pathways, which explains the round shape.
Unlike pneumonia in adults, pneumonia is often initially confined due to immature collateral pathways, which explains the round shape.
 Adenopathy and pleural effusion are rarely seen with round pneumonia.
Adenopathy and pleural effusion are rarely seen with round pneumonia.
SUGGESTED READING
Condon VR. Pneumonia in children. J Thorac Imaging 1991;6:31–44.
Kim Y-W, Donnelly LF. Round pneumonia. Imaging findings in a large series of children. Pediatr Radiol 2007;37:1235–1240.
Yikilmaz A, Lee EY. CT imaging of non-vascular mass-like lesions in children. Pediatr Radiol 2007;37:1253–1263.
GEORGE S. BISSET
HISTORY
A 14-month-old boy with a palpable abdominal mass.
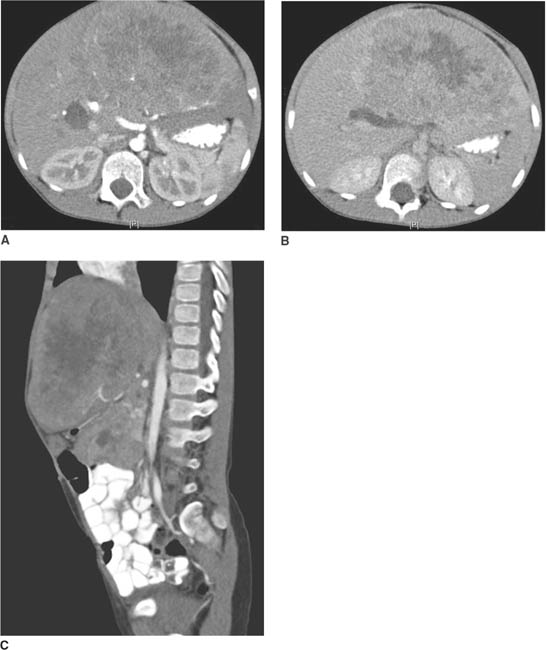
 FIGURE 8-2A, 8-2B, and 8-2C CT of the abdomen during arterial and portal venous phases. A heterogeneously enhancing mass is seen within the left lobe of the liver. The mass displaces the pancreas and IVC posteriorly and displaces the bowel toward the left.
FIGURE 8-2A, 8-2B, and 8-2C CT of the abdomen during arterial and portal venous phases. A heterogeneously enhancing mass is seen within the left lobe of the liver. The mass displaces the pancreas and IVC posteriorly and displaces the bowel toward the left.
DIFFERENTIAL DIAGNOSIS
 Hepatocellular carcinoma: This diagnosis is unlikely because this neoplasm rarely occurs below the age of 2. Furthermore, frequently, there is usually a history of chronic liver disease.
Hepatocellular carcinoma: This diagnosis is unlikely because this neoplasm rarely occurs below the age of 2. Furthermore, frequently, there is usually a history of chronic liver disease.
 Hepatoblastoma: This is the most common malignant primary liver tumor of childhood, usually occurring before the age of 3. The young age of the patient and heterogeneous appearance of the mass in the present case are consistent with this diagnosis.
Hepatoblastoma: This is the most common malignant primary liver tumor of childhood, usually occurring before the age of 3. The young age of the patient and heterogeneous appearance of the mass in the present case are consistent with this diagnosis.
 Mesenchymal hamartoma: This diagnosis is unlikely because it is rare, cystic, and often exophytic, unlike the case illustrated. The diagnosis can be made by prenatal sonography or fetal MR imaging.
Mesenchymal hamartoma: This diagnosis is unlikely because it is rare, cystic, and often exophytic, unlike the case illustrated. The diagnosis can be made by prenatal sonography or fetal MR imaging.
 Hemangioendothelioma: This entity is the most common symptomatic vascular lesion in infancy and most common mesenchymal tumor of childhood. This diagnosis is unlikely in the case illustrated because this benign tumor contains large feeding vessels, which enhance early and areas of variable enhancement. The celiac trunk and hepatic arteries may be enlarged, with tapering of the aorta below the origin of the celiac trunk when this is a high-flow lesion. An infiltrative hemangioendothelioma will demonstrate diffuse nodular enhancement of mulitple masses.
Hemangioendothelioma: This entity is the most common symptomatic vascular lesion in infancy and most common mesenchymal tumor of childhood. This diagnosis is unlikely in the case illustrated because this benign tumor contains large feeding vessels, which enhance early and areas of variable enhancement. The celiac trunk and hepatic arteries may be enlarged, with tapering of the aorta below the origin of the celiac trunk when this is a high-flow lesion. An infiltrative hemangioendothelioma will demonstrate diffuse nodular enhancement of mulitple masses.
 Hepatic adenoma: These tumors are extremely rare in young children. Furthermore, the lesion may contains regions of fatty change or hemorrhage. This finding is not seen in the case illustrated, further making this diagnosis an unlikely one.
Hepatic adenoma: These tumors are extremely rare in young children. Furthermore, the lesion may contains regions of fatty change or hemorrhage. This finding is not seen in the case illustrated, further making this diagnosis an unlikely one.
DIAGNOSIS
Hepatoblastoma
KEY FACTS
Clinical
 Hepatoblastoma is probably an infantile form of hepa-tocellular carcinoma.
Hepatoblastoma is probably an infantile form of hepa-tocellular carcinoma.
 This tumor primarily occurs before the age of 3 (median age: 1 year).
This tumor primarily occurs before the age of 3 (median age: 1 year).
 A male-to-female predominance between 2:1 and 3:1 has been reported.
A male-to-female predominance between 2:1 and 3:1 has been reported.
 The tumor is associated with Beckwith-Wiedemann syndrome (i.e., prematurity, hemihypertrophy and biliary atresia).
The tumor is associated with Beckwith-Wiedemann syndrome (i.e., prematurity, hemihypertrophy and biliary atresia).
 Nearly all patients with this tumor have elevated serum levels of alpha-fetoprotein.
Nearly all patients with this tumor have elevated serum levels of alpha-fetoprotein.
 Prognosis is usually good if there are no metastases and the tumor is limited to the liver and is completely resectable.
Prognosis is usually good if there are no metastases and the tumor is limited to the liver and is completely resectable.
Radiologic
 Hepatoblastoma usually presents as a single mass. However, multifocal involvement can sometimes occur.
Hepatoblastoma usually presents as a single mass. However, multifocal involvement can sometimes occur.
 On contrast-enhanced CT examination or MR imaging, the mass usually demonstrates heterogeneous enhancement.
On contrast-enhanced CT examination or MR imaging, the mass usually demonstrates heterogeneous enhancement.
 Periportal areas of low attenuation, as seen in this case, or signal abnormality are likely related to tumor invasion of periportal lymphatics. Tumoral calcification occurs in approximately 40%.
Periportal areas of low attenuation, as seen in this case, or signal abnormality are likely related to tumor invasion of periportal lymphatics. Tumoral calcification occurs in approximately 40%.
 Splaying of hepatic veins around the mass can be seen occasionally.
Splaying of hepatic veins around the mass can be seen occasionally.
 The sonographic appearance is that of a hypoechoic mass with displacement of the hepatic arterial and portal venous vessels by the tumor.
The sonographic appearance is that of a hypoechoic mass with displacement of the hepatic arterial and portal venous vessels by the tumor.
SUGGESTED READING
Boechat MI, Kangarloo H, Ortega J, et al. Primary liver tumors in children: comparison of CT and MR imaging. Radiology 1988;169:727–732.
Finegold MJ, Egler RA, et al. Liver tumors: pediatric population. Liver Transpl 2008;14:1545–1556.
Finn JP, Hall-Craggs MA, Dicks-Mireaux C, et al. Primary malignant liver tumors in childhood: assessment of resectability with high field MR and comparison with CT. Pediatr Radiol 1990;21:34–38.
Jabra AA, Fishman EK, Taylor GA. Hepatic masses in infants and children. CT evaluation. AJR Am J Roentgenol 1992;158: 143–149.
Pobiel RS, Bisset GB. Pictorial essay: imaging of liver tumors in the infant and child. Pediatr Radiol 1995;25:495–506.
Siegel MJ. Pediatric liver imaging. Semin Liv Dis 2001;21:251–269.
DONALD P. FRUSH AND SARA M. O’HARA
HISTORY
A toddler with urinary retention, constipation, and distended abdomen.
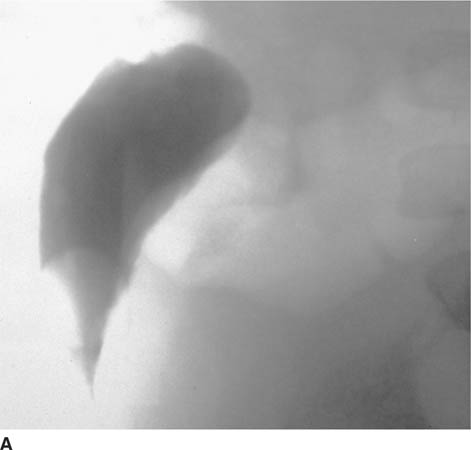
 FIGURE 8-3A Lateral spot film from voiding cystoure-throgram. The bladder neck is elongated, and the entire bladder is displaced upward and anteriorly out of the pelvis.
FIGURE 8-3A Lateral spot film from voiding cystoure-throgram. The bladder neck is elongated, and the entire bladder is displaced upward and anteriorly out of the pelvis.
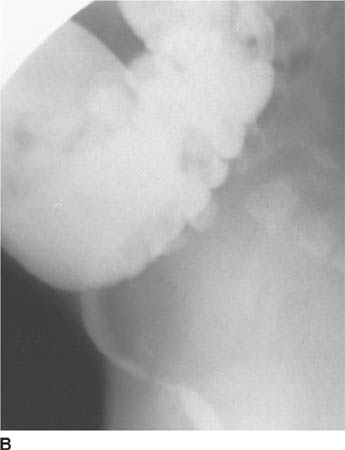
 FIGURE 8-3B Lateral spot film from barium enema examination. The rectosigmoid is compressed and displaced anteriorly. No bowel gas or stool is visible in the presacral space.
FIGURE 8-3B Lateral spot film from barium enema examination. The rectosigmoid is compressed and displaced anteriorly. No bowel gas or stool is visible in the presacral space.
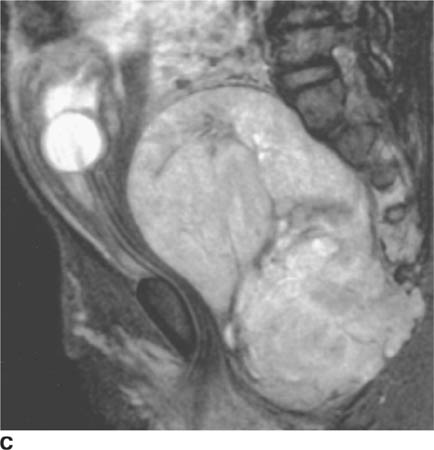
 FIGURE 8-3C Sagittal T2-weighted fast spin echo MRI with fat saturation. A mixed-signal intensity mass is present in the presacral region, extending between the sacral segments and into the posterior musculature. The Foley catheter within the bladder is displaced anteriorly and superiorly.
FIGURE 8-3C Sagittal T2-weighted fast spin echo MRI with fat saturation. A mixed-signal intensity mass is present in the presacral region, extending between the sacral segments and into the posterior musculature. The Foley catheter within the bladder is displaced anteriorly and superiorly.
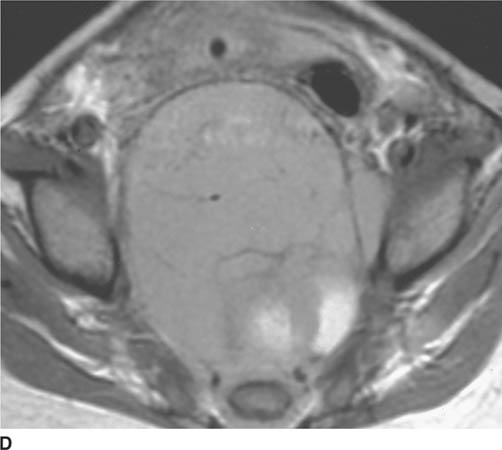
 FIGURE 8-3D Axial unenhanced T1-weighted MRI. A predominantly intermediate-signal intensity soft tissue mass is seen encasing the lower sacrum and displacing the rectum anteriorly and to the left.
FIGURE 8-3D Axial unenhanced T1-weighted MRI. A predominantly intermediate-signal intensity soft tissue mass is seen encasing the lower sacrum and displacing the rectum anteriorly and to the left.
 Ovarian teratoma: Any tumorous growth from the ovary could grow to the size shown in this case. However, extension around the sacrum is not a feature seen in ovarian tumor, making this an unlikely diagnosis.
Ovarian teratoma: Any tumorous growth from the ovary could grow to the size shown in this case. However, extension around the sacrum is not a feature seen in ovarian tumor, making this an unlikely diagnosis.
 Abscess: This entity cannot be excluded on the basis of imaging findings alone. However, it is an unlikely diagnosis because the clinical history does not suggest the presence of infectious process.
Abscess: This entity cannot be excluded on the basis of imaging findings alone. However, it is an unlikely diagnosis because the clinical history does not suggest the presence of infectious process.
 Neuroblastoma (or other ganglion tumor such as ganglioneuroma): The location and appearance of the mass are consistent with this diagnosis. However, posterior encasement and direct sacral involvement are not findings that would be expected in pelvic neuroblastoma.
Neuroblastoma (or other ganglion tumor such as ganglioneuroma): The location and appearance of the mass are consistent with this diagnosis. However, posterior encasement and direct sacral involvement are not findings that would be expected in pelvic neuroblastoma.
 Sacrococcygeal germ cell tumor (i.e., teratoma): Ter-atoma is the best diagnosis, based on the presacral and retrorectal location and, in particular, the encasement of sacral segments.
Sacrococcygeal germ cell tumor (i.e., teratoma): Ter-atoma is the best diagnosis, based on the presacral and retrorectal location and, in particular, the encasement of sacral segments.
 Anterior meningocele: This diagnosis is unlikely because the mass is not cystic.
Anterior meningocele: This diagnosis is unlikely because the mass is not cystic.
 Rhabdomyosarcoma: Because posterior sacral extension would be unusual for rhabdomyosarcoma, this is an unlikely diagnosis.
Rhabdomyosarcoma: Because posterior sacral extension would be unusual for rhabdomyosarcoma, this is an unlikely diagnosis.
 Primary bone tumor: A primary bone tumor is not a likely consideration because there is no bony destruction and sacral involvement in this case is relatively minimal. Primary sacral bone tumors are extremely rare at this age.
Primary bone tumor: A primary bone tumor is not a likely consideration because there is no bony destruction and sacral involvement in this case is relatively minimal. Primary sacral bone tumors are extremely rare at this age.
 Gastrointestinal duplication: This diagnosis is unlikely because rectal duplications are very rare and extension to sacrum would not be expected.
Gastrointestinal duplication: This diagnosis is unlikely because rectal duplications are very rare and extension to sacrum would not be expected.
DIAGNOSIS
Malignant sacrococcygeal germ cell tumor (teratoma)
KEY FACTS
Clinical
 Sacrococcygeal teratomas are relatively rare, with an estimated prevalence of 1 in 35,000 to 40,000 births.
Sacrococcygeal teratomas are relatively rare, with an estimated prevalence of 1 in 35,000 to 40,000 births.
 Sacrococcygeal teratoma is the most common solid tumor in neonates.
Sacrococcygeal teratoma is the most common solid tumor in neonates.
 Eighty percent are diagnosed before 6 months of age.
Eighty percent are diagnosed before 6 months of age.
 Seventy-five percent are in females; the gender ratio for malignant lesions is equal, however.
Seventy-five percent are in females; the gender ratio for malignant lesions is equal, however.
 The category of germ cell tumors includes teratomas, choriocarcinomas, embryonal cell carcinomas, yolk sac tumors, and mixed types.
The category of germ cell tumors includes teratomas, choriocarcinomas, embryonal cell carcinomas, yolk sac tumors, and mixed types.
 Teratomas are the most common sacrococcygeal tumors in children.
Teratomas are the most common sacrococcygeal tumors in children.
 Teratomas are classified as mature, immature, or malignant. Malignancy is rare in mature types. Overall 60% of teratomes are benign.
Teratomas are classified as mature, immature, or malignant. Malignancy is rare in mature types. Overall 60% of teratomes are benign.
 Treatment consists of chemotherapy and surgical resection.
Treatment consists of chemotherapy and surgical resection.
 Early diagnosis and surgical excision is indicated because the incidence of malignant transformation increases with advancing age.
Early diagnosis and surgical excision is indicated because the incidence of malignant transformation increases with advancing age.
 Four types of sacral teratomas have been described, according to the internal and external components of the mass:
Four types of sacral teratomas have been described, according to the internal and external components of the mass:
 Type 1: primarily external to the pelvis (50% are Type 1)
Type 1: primarily external to the pelvis (50% are Type 1)
 Type 2: predominantly external to the pelvis with intra-pelvic component
Type 2: predominantly external to the pelvis with intra-pelvic component
 Type 3: primarily intrapelvic with only a small extra-pelvic portion
Type 3: primarily intrapelvic with only a small extra-pelvic portion
 Type 4: entirely presacral without extrapelvic extension
Type 4: entirely presacral without extrapelvic extension
 In general, the greater the intrapelvic component, the greater the likelihood of malignancy.
In general, the greater the intrapelvic component, the greater the likelihood of malignancy.
Radiologic
 Calcification is visible by CT in 50% to 60% of sacrococcygeal teratomas and is more frequent in benign lesions. Amorphous, chunky, punctate, and spiculated calcifications have all been described.
Calcification is visible by CT in 50% to 60% of sacrococcygeal teratomas and is more frequent in benign lesions. Amorphous, chunky, punctate, and spiculated calcifications have all been described.
 Cystic and fatty components can also be seen within the mass.
Cystic and fatty components can also be seen within the mass.
 Primarily solid lesions are more likely to be malignant, and those containing fat and calcification with cystic regions are more likely benign; however, exceptions exist.
Primarily solid lesions are more likely to be malignant, and those containing fat and calcification with cystic regions are more likely benign; however, exceptions exist.
 Direct invasion of adjacent structures suggests malignant transformation.
Direct invasion of adjacent structures suggests malignant transformation.
 Elevated serum alpha-fetoprotein levels are often present and are frequently followed in conjunction with imaging findings as a measure of tumor response.
Elevated serum alpha-fetoprotein levels are often present and are frequently followed in conjunction with imaging findings as a measure of tumor response.
Presacral masses (usually anterior meningocele or germ cell tumor) can coexist with two other entities: anorectal malformations and sacral anomalies, a constellation that is referred to as Currarino’s triad, or the ASP (anorectal malformation, sacral anomalies and presacral mass) triad or ASP complex (anorectal malformation, sacrococcygeal osseous defect, and presacral mass).
SUGGESTED READING
Kocaoglu M, Frush DP. Pediatric presacral masses. Radiographics 2006;26:833–857.
Siegel MJ. Pelvic tumors in childhood. Radiol Clin North Am 1997;35: 1455–1475.
DONALD P. FRUSH
HISTORY
A 5-year-old boy with several months of increasing right knee pain and limping.
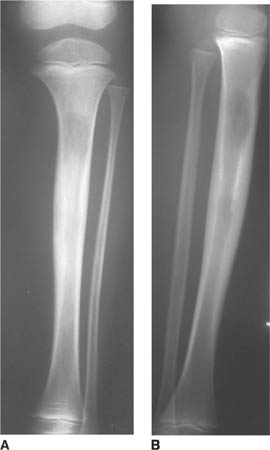
 FIGURES 8-4A and 8-4B Frontal (A) and lateral (B) radiographs of the right tibia and fibula. An ill-defined metaphy-seal lesion is present that demonstrates mixed sclerosis and lucency at the anterior aspect of the tibia.
FIGURES 8-4A and 8-4B Frontal (A) and lateral (B) radiographs of the right tibia and fibula. An ill-defined metaphy-seal lesion is present that demonstrates mixed sclerosis and lucency at the anterior aspect of the tibia.
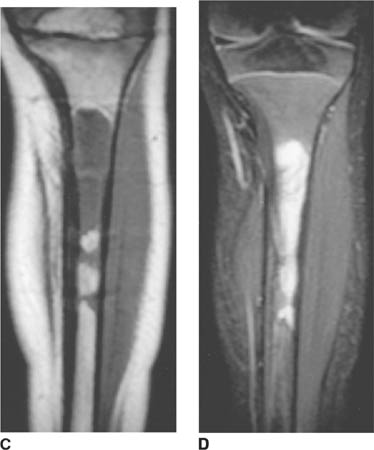
 FIGURES 8-4 C and 8-4D Unenhanced coronal T1-weighted (C) and T2-weighted (D) images of the right tibia. Abnormal low T1, high T2 marrow signal is present in the tibial metaphysis, extending into the diaphysis, with destruction of the cortex. Extraosseous tumor and edema are demonstrated on the T2-weighted image.
FIGURES 8-4 C and 8-4D Unenhanced coronal T1-weighted (C) and T2-weighted (D) images of the right tibia. Abnormal low T1, high T2 marrow signal is present in the tibial metaphysis, extending into the diaphysis, with destruction of the cortex. Extraosseous tumor and edema are demonstrated on the T2-weighted image.
 The major considerations in this differential are the small, round, blue cell tumors of childhood. All may present with a permeative pattern and a wide zone of transition.
The major considerations in this differential are the small, round, blue cell tumors of childhood. All may present with a permeative pattern and a wide zone of transition.
 Ewing sarcoma of bone (or Ewing sarcoma family, including primitive neuroectodermal tumor or PNET, Askin’s tumor, and extraosseous Ewing’s sarcoma): A likely consideration based on the history and radiographic findings.
Ewing sarcoma of bone (or Ewing sarcoma family, including primitive neuroectodermal tumor or PNET, Askin’s tumor, and extraosseous Ewing’s sarcoma): A likely consideration based on the history and radiographic findings.
 Metastases: Considerations are especially those due to neuroblastoma and rhabdomyosarcoma. With rhabdo-myosarcoma, presenting symptoms are usually due to the primary mass rather than metastatic disease. The imaging findings in this case could be due to neuroblastoma metastasis.
Metastases: Considerations are especially those due to neuroblastoma and rhabdomyosarcoma. With rhabdo-myosarcoma, presenting symptoms are usually due to the primary mass rather than metastatic disease. The imaging findings in this case could be due to neuroblastoma metastasis.
 Leukemia or lymphoma: This is also a reasonable consideration because the lymphoproliferative malignancies are the most common childhood cancer. Bone abnormalities in leukemia are fairly common (osteoporosis, metaphyseal lucencies, periosteal reaction, sclerotic or lucent lesions, permeative pattern); the involvement is most often multifocal and symmetrical, however, making the appearance in this case unusual. Lymphoma may have an identical appearance. However, primary lymphoma of bone is much less likely than Ewing’s sarcoma.
Leukemia or lymphoma: This is also a reasonable consideration because the lymphoproliferative malignancies are the most common childhood cancer. Bone abnormalities in leukemia are fairly common (osteoporosis, metaphyseal lucencies, periosteal reaction, sclerotic or lucent lesions, permeative pattern); the involvement is most often multifocal and symmetrical, however, making the appearance in this case unusual. Lymphoma may have an identical appearance. However, primary lymphoma of bone is much less likely than Ewing’s sarcoma.
Considerations other than the small, round, blue cell tumors include:
 Osteomyelitis: It may be impossible to differentiate osteomyelitis from Ewing’s sarcoma clinically and radiographically.
Osteomyelitis: It may be impossible to differentiate osteomyelitis from Ewing’s sarcoma clinically and radiographically.
 Eosinophilic granuloma: This entity has a large variety of appearances (classically a lucent defect) that include an aggressive appearance such as this one.
Eosinophilic granuloma: This entity has a large variety of appearances (classically a lucent defect) that include an aggressive appearance such as this one.
DIAGNOSIS
Ewing sarcoma
KEY FACTS
Clinical
 Although originally described as separate entities, Ewing’s sarcoma, primitive neuroectodermal tumors (PNET), Askin’s tumors, adult neuroblastoma, and soft tissue Ewing’s sarcoma are all part of a spectrum of neoplastic diseases called the Ewing’s sarcoma family of tumors (EFT).
Although originally described as separate entities, Ewing’s sarcoma, primitive neuroectodermal tumors (PNET), Askin’s tumors, adult neuroblastoma, and soft tissue Ewing’s sarcoma are all part of a spectrum of neoplastic diseases called the Ewing’s sarcoma family of tumors (EFT).
 Classic Ewing’s sarcoma occurs most commonly in the second half of the first decade of life and the first half of the second decade, a slightly younger age distribution than that of osteosarcoma.
Classic Ewing’s sarcoma occurs most commonly in the second half of the first decade of life and the first half of the second decade, a slightly younger age distribution than that of osteosarcoma.
 Ewing sarcoma affects males slightly more commonly than females (3:2), and is uncommon in Asians and African Americans.
Ewing sarcoma affects males slightly more commonly than females (3:2), and is uncommon in Asians and African Americans.
 Pain, swelling, or both at the site of the primary tumor are the most common presenting symptoms in Ewing sarcoma. However, the presentation may be similar to that of osteomyelitis, with systemic symptoms of fever, elevated sedimentation rate and leukocytosis.
Pain, swelling, or both at the site of the primary tumor are the most common presenting symptoms in Ewing sarcoma. However, the presentation may be similar to that of osteomyelitis, with systemic symptoms of fever, elevated sedimentation rate and leukocytosis.
 Metastatic spread of Ewing sarcoma occurs equally to the lungs and distant bones, and is found in approximately 25% of cases at the time of diagnosis. Subclinical metastasis is thought to be present in the majority of patients because of the high rate of relapse in patients treated only with local management.
Metastatic spread of Ewing sarcoma occurs equally to the lungs and distant bones, and is found in approximately 25% of cases at the time of diagnosis. Subclinical metastasis is thought to be present in the majority of patients because of the high rate of relapse in patients treated only with local management.
 Poor prognostic indicators include large size (> 8 cm), central location, unresectability older age, and elevated erythrocyte sedimentation rate as well as leukocyte count at the time of presentation.
Poor prognostic indicators include large size (> 8 cm), central location, unresectability older age, and elevated erythrocyte sedimentation rate as well as leukocyte count at the time of presentation.
Radiologic
 Ewing sarcoma can develop in almost any bone in the body, and equally affects both long and flat bones.
Ewing sarcoma can develop in almost any bone in the body, and equally affects both long and flat bones.
 Lesions usually occur in the midshaft of a long bone, rather than the ends as with osteosarcoma.
Lesions usually occur in the midshaft of a long bone, rather than the ends as with osteosarcoma.
 Plain films underestimate the extent of bone involvement. The most typical pattern is a permeative, lytic pattern with associated periosteal reaction. However, other patterns include larger, “moth-eaten” lytic foci, a mixed lytic and sclerotic pattern, bony expansion, and a predominantly sclerotic pattern.
Plain films underestimate the extent of bone involvement. The most typical pattern is a permeative, lytic pattern with associated periosteal reaction. However, other patterns include larger, “moth-eaten” lytic foci, a mixed lytic and sclerotic pattern, bony expansion, and a predominantly sclerotic pattern.
 Ewing’s sarcoma of flat bones typically has a large soft tissue component relative to the osseous component, in contrast to Ewing’s sarcoma of long bones.
Ewing’s sarcoma of flat bones typically has a large soft tissue component relative to the osseous component, in contrast to Ewing’s sarcoma of long bones.
 MR imaging is the imaging modality of choice to determine tumor size, intra- and extraosseous extension, involvement of adjacent organs and neurovascular structures. Intravenous contrast material can be used to distinguish tumor from peritumoral edema.
MR imaging is the imaging modality of choice to determine tumor size, intra- and extraosseous extension, involvement of adjacent organs and neurovascular structures. Intravenous contrast material can be used to distinguish tumor from peritumoral edema.
 MR imaging of the entire bone at the primary site of involvement is important as skip metastases occur with Ewing’s sarcoma as well as osteosarcoma.
MR imaging of the entire bone at the primary site of involvement is important as skip metastases occur with Ewing’s sarcoma as well as osteosarcoma.
 The assessment for metastatic disease should include a chest CT to search for pulmonary metastatic disease and a bone scan for distant bony metastases.
The assessment for metastatic disease should include a chest CT to search for pulmonary metastatic disease and a bone scan for distant bony metastases.
SUGGESTED READING
Bielack SS, Carrle D. State-of-the-art approach in selective curable tumors: bone sarcoma. Ann Oncol 2008;19: vii155–vii160.
Grier HE. The Ewing family of tumors: Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatr Clin N Am 1997;44(4):991–1004.
Meyer JS, Mackenzie W. Malignant bone tumors and limb-salvage surgery in children. Pediatr Radiol 2004;34:606–613.
CHARLES MAXFIELD
HISTORY
A 6-year-old girl with acute stridor, high fever, and respiratory distress.
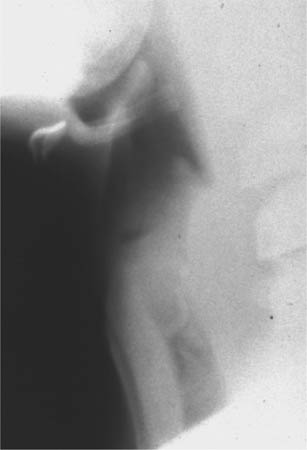
 FIGURE 8-5 A lateral airway radiograph shows subtle heterogeneous haziness (i.e., so-called clouding) in the cervical segment of the trachea, along with a curvilinear density in the subglottic trachea which represents an exudative plaque.
FIGURE 8-5 A lateral airway radiograph shows subtle heterogeneous haziness (i.e., so-called clouding) in the cervical segment of the trachea, along with a curvilinear density in the subglottic trachea which represents an exudative plaque.
 Tracheal foreign body: This diagnosis is a possible consideration but there is no history of aspiration of a foreign body. Furthermore, high fever would not be expected, and the age of patients with aspirated foreign bodies is usually much younger than the patient shown here. This diagnosis is unlikely.
Tracheal foreign body: This diagnosis is a possible consideration but there is no history of aspiration of a foreign body. Furthermore, high fever would not be expected, and the age of patients with aspirated foreign bodies is usually much younger than the patient shown here. This diagnosis is unlikely.
 Papilloma: Papillomas usually have the clinical presentation of chronic hoarseness, rather than the acute stridor and fever found in the patient presented here. Furthermore, the opacities in the radiographs shown in this case are not mass-like, as would be expected with a papilloma. This diagnosis is an unlikely one.
Papilloma: Papillomas usually have the clinical presentation of chronic hoarseness, rather than the acute stridor and fever found in the patient presented here. Furthermore, the opacities in the radiographs shown in this case are not mass-like, as would be expected with a papilloma. This diagnosis is an unlikely one.
 Tracheal mucus: Mucus within the trachea can produce intraluminal opacities on radiographs. However, the presence of tracheal mucus would not explain the toxic appearance of the patient or the tracheal narrowing and clouding in the case shown here.
Tracheal mucus: Mucus within the trachea can produce intraluminal opacities on radiographs. However, the presence of tracheal mucus would not explain the toxic appearance of the patient or the tracheal narrowing and clouding in the case shown here.
 Epiglottitis: The history presented in this case is very suggestive of epiglottitis but the epiglottis in the radiographs is normal in appearance, making this diagnosis unlikely.
Epiglottitis: The history presented in this case is very suggestive of epiglottitis but the epiglottis in the radiographs is normal in appearance, making this diagnosis unlikely.
 Laryngotracheobronchitis: This entity, also known as croup, usually occurs in children between the ages of 6 months and 3 years, i.e., at an age that is much younger than the child presented here. Furthermore, narrowing of the subglottic trachea (the so-called steeple sign) typical of croup is not present. This diagnosis is incorrect.
Laryngotracheobronchitis: This entity, also known as croup, usually occurs in children between the ages of 6 months and 3 years, i.e., at an age that is much younger than the child presented here. Furthermore, narrowing of the subglottic trachea (the so-called steeple sign) typical of croup is not present. This diagnosis is incorrect.
 Granuloma: This entity is typically seen following unlikely previous tracheal intubation or other source of trauma to the trachea. No such history is given in the case shown here, making this diagnosis unlikely.
Granuloma: This entity is typically seen following unlikely previous tracheal intubation or other source of trauma to the trachea. No such history is given in the case shown here, making this diagnosis unlikely.
 Bacterial tracheitis: This entity is also known as membranous croup or membranous laryngotracheobronchi-tis. Children with this diagnosis typically have an acute, toxic presentation, such as in the case shown here. Furthermore, on radiographs, bacterial tracheitis typically is seen as subglottic tracheal narrowing in association with intraluminal plaque-like opacities, as in the images shown here. This diagnosis is correct.
Bacterial tracheitis: This entity is also known as membranous croup or membranous laryngotracheobronchi-tis. Children with this diagnosis typically have an acute, toxic presentation, such as in the case shown here. Furthermore, on radiographs, bacterial tracheitis typically is seen as subglottic tracheal narrowing in association with intraluminal plaque-like opacities, as in the images shown here. This diagnosis is correct.
 Retropharyngeal abscess: The retropharyngeal soft tissues are normal in the images shown here, thereby excluding the diagnosis of a retropharyngeal abscess as a cause of the stridor.
Retropharyngeal abscess: The retropharyngeal soft tissues are normal in the images shown here, thereby excluding the diagnosis of a retropharyngeal abscess as a cause of the stridor.
DIAGNOSIS
Bacterial tracheitis
KEY FACTS
Clinical
 Tracheitis is a rare entity compared to other causes of respiratory difficulty in children, such as croup and ret-ropharyngeal abscess.
Tracheitis is a rare entity compared to other causes of respiratory difficulty in children, such as croup and ret-ropharyngeal abscess.
 Acute onset of stridor is the typical clinical presentation of bacterial tracheitis.
Acute onset of stridor is the typical clinical presentation of bacterial tracheitis.
 The mean age at presentation is 4 years old, but bacterial tracheitis can be seen at any age.
The mean age at presentation is 4 years old, but bacterial tracheitis can be seen at any age.
 Prompt recognition is important because membranes can develop relatively quickly and acutely obstruct the airway.
Prompt recognition is important because membranes can develop relatively quickly and acutely obstruct the airway.
 Infection is bacterial in origin. Staphyloccus aureus is the most common agent, but occasionally Streptococcus pneumoniae and Haemophilus influenza can produce the infection. However, tracheitis can also occur as a superinfection following a viral upper airway process.
Infection is bacterial in origin. Staphyloccus aureus is the most common agent, but occasionally Streptococcus pneumoniae and Haemophilus influenza can produce the infection. However, tracheitis can also occur as a superinfection following a viral upper airway process.
 If clinical or radiographic findings suggest tracheitis, flexible endoscopy is indicated to confirm the diagnosis.
If clinical or radiographic findings suggest tracheitis, flexible endoscopy is indicated to confirm the diagnosis.
 Management primarily includes supportive care (e.g., humidification, antibiotics, and suctioning), but “elective” intubation may be necessary to protect the airway from obstruction.
Management primarily includes supportive care (e.g., humidification, antibiotics, and suctioning), but “elective” intubation may be necessary to protect the airway from obstruction.
Radiologic
 Lateral airway radiographs often show an indistinct appearance (i.e., so-called clouding) of the trachea, intraluminal tracheal opacities, and tracheal wall irregularity.
Lateral airway radiographs often show an indistinct appearance (i.e., so-called clouding) of the trachea, intraluminal tracheal opacities, and tracheal wall irregularity.
 Occasionally, associated subglottic narrowing can be present.
Occasionally, associated subglottic narrowing can be present.
 The epiglottis and retropharyngeal tissues are normal in tracheitis, distinguishing this entity from other infectious causes of acute stridor in children.
The epiglottis and retropharyngeal tissues are normal in tracheitis, distinguishing this entity from other infectious causes of acute stridor in children.
 Half of cases have associated pneumonia.
Half of cases have associated pneumonia.
 A normal radiograph does not exclude the diagnosis of tracheitis.
A normal radiograph does not exclude the diagnosis of tracheitis.
 The major alternative diagnosis based on radiographic findings is tracheal mucus accumulation. The two entities can be indistinguishable on the lateral airway radiograph. Because early treatment of tracheitis is important, this diagnosis should be favored in equivocal cases and treatment should begin.
The major alternative diagnosis based on radiographic findings is tracheal mucus accumulation. The two entities can be indistinguishable on the lateral airway radiograph. Because early treatment of tracheitis is important, this diagnosis should be favored in equivocal cases and treatment should begin.
SUGGESTED READING
Gallagher P, Myer CM. An approach to the diagnosis and treatment of membranous laryngotracheobronchitis in infants and children. Pediatr Emerg Care 1991;7:337–342.
Hopkins A, Lahiri T, Salerno R, Heath B. Changing epidemiology of life-threatening upper airway infections: the reemergence of bacterial tracheitis. Pediatrics 2006;118:1418–1421.
John SD, Swischuk LE. Stridor and upper airway obstruction in infants and children. Radiographics 1991;12:625–643.
Loftis L. Acute infectious upper airway obstruction in children. Semin Pediatr Infect Dis 2006;17:5–10.
Seigler R. Bacterial tracheitis: recognition and treatment. J SC Med Assoc 1993;89:83–87.
CAROLINE L. HOLLINGSWORTH AND SARA M. O’HARA
HISTORY
A 5-week-old girl with persistent nonbilious vomiting but with no history of diarrhea or fever.
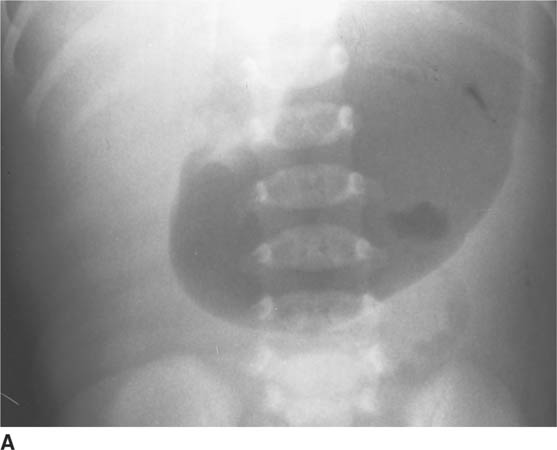
 FIGURE 8-6A Anteroposterior abdominal radiograph. The stomach is markedly distended. A large amount of gas is present in the stomach but only a minimal amount of gas is seen in the small bowel.
FIGURE 8-6A Anteroposterior abdominal radiograph. The stomach is markedly distended. A large amount of gas is present in the stomach but only a minimal amount of gas is seen in the small bowel.
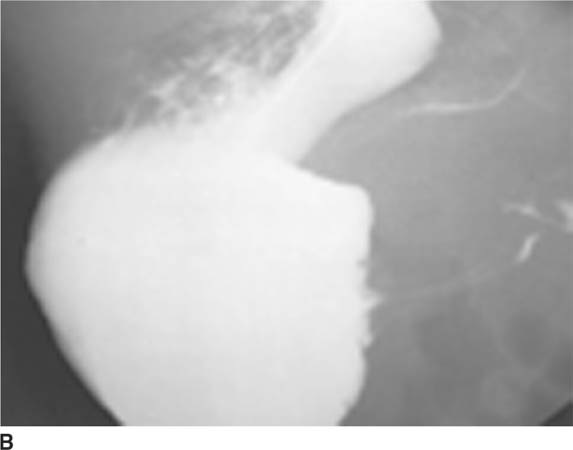
 FIGURE 8-6B Upper gastrointestinal series; lateral view of the stomach. A large pool of barium is present in the stomach. Thin lines of barium are visible in an elongated pylorus. Only a very small amount of barium is present in the duodenal bulb.
FIGURE 8-6B Upper gastrointestinal series; lateral view of the stomach. A large pool of barium is present in the stomach. Thin lines of barium are visible in an elongated pylorus. Only a very small amount of barium is present in the duodenal bulb.
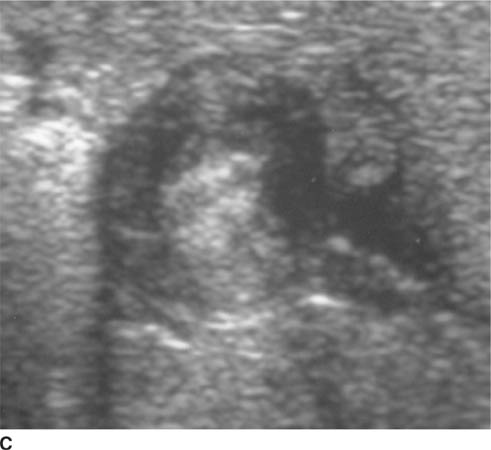
 FIGURE 8-6C Transverse sonographic view of the right upper quadrant. The hypoechoic muscularis is thickened and surrounds echogenic redundant mucosa.
FIGURE 8-6C Transverse sonographic view of the right upper quadrant. The hypoechoic muscularis is thickened and surrounds echogenic redundant mucosa.
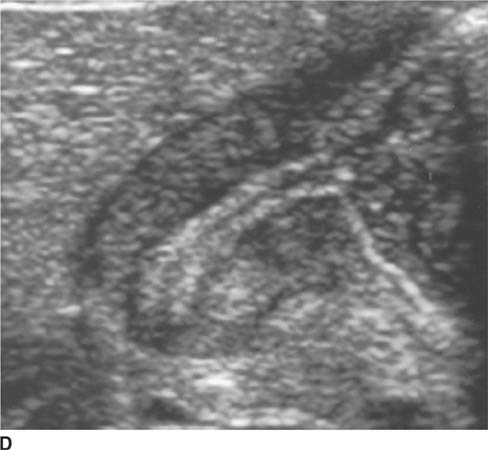
 FIGURE 8-6D Longitudinal sonographic view of the right upper quadrant. The pyloric channel is elongated and has a thickened muscularis.
FIGURE 8-6D Longitudinal sonographic view of the right upper quadrant. The pyloric channel is elongated and has a thickened muscularis.
 Gastroesophageal reflux: The patient’s history makes this diagnosis a possibility. However, no reflux of contrast material is seen at fluoroscopy in Figure 8-6B. Instead, a pyloric abnormality is visible on upper gastrointestinal series and sonography.
Gastroesophageal reflux: The patient’s history makes this diagnosis a possibility. However, no reflux of contrast material is seen at fluoroscopy in Figure 8-6B. Instead, a pyloric abnormality is visible on upper gastrointestinal series and sonography.
 Pylorospasm: Pylorospasm can be seen in patients who are extremely agitated, dehydrated, or septic, unlike the patient discussed here. Furthermore, pylorospasm is unlikely to persist throughout the entire series of imaging examinations shown here but would, instead, be expected to be transitory. Thus, this diagnosis is an unlikely one based on the clinical history and imaging findings.
Pylorospasm: Pylorospasm can be seen in patients who are extremely agitated, dehydrated, or septic, unlike the patient discussed here. Furthermore, pylorospasm is unlikely to persist throughout the entire series of imaging examinations shown here but would, instead, be expected to be transitory. Thus, this diagnosis is an unlikely one based on the clinical history and imaging findings.
 Pyloric channel ulcer: This entity could account for the delayed gastric emptying and vomiting. However, no ulcer is identified on the imaging studies shown; furthermore, this entity usually occurs at an older age.
Pyloric channel ulcer: This entity could account for the delayed gastric emptying and vomiting. However, no ulcer is identified on the imaging studies shown; furthermore, this entity usually occurs at an older age.
 Antral web: Antral web is another cause of delayed gastric emptying and vomiting. However, no evidence of an obstructing web is seen on the imaging studies, making this an incorrect diagnosis.
Antral web: Antral web is another cause of delayed gastric emptying and vomiting. However, no evidence of an obstructing web is seen on the imaging studies, making this an incorrect diagnosis.
 Hypertrophic pyloric stenosis (HPS): The sonographic findings of concentric thickening of the pyloric muscu-laris and elongation of the pyloric channel, seen in the case shown here, are typical for this entity. The multiple thin tracks of barium visible in the pyloric channel on the UGI series represent folds of mucosa in a narrowed channel, further evidence of HPS. This entity is the best diagnosis based on the history and imaging findings.
Hypertrophic pyloric stenosis (HPS): The sonographic findings of concentric thickening of the pyloric muscu-laris and elongation of the pyloric channel, seen in the case shown here, are typical for this entity. The multiple thin tracks of barium visible in the pyloric channel on the UGI series represent folds of mucosa in a narrowed channel, further evidence of HPS. This entity is the best diagnosis based on the history and imaging findings.
 Malrotation with obstruction: In the first month of life the obstruction from malrotation is due to midgut volvulus. With malrotation later in infancy and childhood, peritoneal reflections (so-called Ladd bands) cross the duodenum and fix the malpositioned cecum, causing obstruction and vomiting. Thus, in malrotation the site of obstruction is the duodenum, not the pylorus (as in the case shown here). In addition, volvulus typically presents with bilious vomiting; nonbilious vomiting is rare. This diagnosis is incorrect.
Malrotation with obstruction: In the first month of life the obstruction from malrotation is due to midgut volvulus. With malrotation later in infancy and childhood, peritoneal reflections (so-called Ladd bands) cross the duodenum and fix the malpositioned cecum, causing obstruction and vomiting. Thus, in malrotation the site of obstruction is the duodenum, not the pylorus (as in the case shown here). In addition, volvulus typically presents with bilious vomiting; nonbilious vomiting is rare. This diagnosis is incorrect.
 Duodenal atresia: Congenital bowel atresia (e.g., duodenal atresia and ileal atresia) clinically presents at a much younger age than that of the patient in the case illustrated. Duodenal atresia is evident at birth with a radiographic “double bubble” due to gas-distended stomach and proximal duodenum. Ileal atresia is also evident at birth with distension and vomiting, which is often bilious. This diagnosis is incorrect.
Duodenal atresia: Congenital bowel atresia (e.g., duodenal atresia and ileal atresia) clinically presents at a much younger age than that of the patient in the case illustrated. Duodenal atresia is evident at birth with a radiographic “double bubble” due to gas-distended stomach and proximal duodenum. Ileal atresia is also evident at birth with distension and vomiting, which is often bilious. This diagnosis is incorrect.
DIAGNOSIS
Hypertrophic pyloric stenosis
KEY FACTS
Clinical
 The clinical presentation is that of nonbilious vomiting, which gradually progresses in severity over time from low-velocity regurgitation to projectile vomiting. Weight loss, dehydration, and hypochloremic acidosis can occur secondarily. Most patients appear healthy but hungry.
The clinical presentation is that of nonbilious vomiting, which gradually progresses in severity over time from low-velocity regurgitation to projectile vomiting. Weight loss, dehydration, and hypochloremic acidosis can occur secondarily. Most patients appear healthy but hungry.
 HPS is characterized by thickening of the circular muscular layer of the pyloric canal, which results in narrowing of the pyloric channel.
HPS is characterized by thickening of the circular muscular layer of the pyloric canal, which results in narrowing of the pyloric channel.
 HPS is the most common condition necessitating abdominal surgery in the infant and most common cause of gastric outlet obstruction in the first few weeks of life.
HPS is the most common condition necessitating abdominal surgery in the infant and most common cause of gastric outlet obstruction in the first few weeks of life.
 HPS typically presents in Caucasian boys between the ages of 3 weeks and 8 weeks.
HPS typically presents in Caucasian boys between the ages of 3 weeks and 8 weeks.
 A palpable thickened pylorus (i.e., so-called olive) in the epigastrium is pathognomonic but is usually detectable only by a trained examiner. When this finding is present in the setting of suspected HPS, no preoperative imaging is necessary. Nonetheless, in most centers a pyloric sonogram will be obtained.
A palpable thickened pylorus (i.e., so-called olive) in the epigastrium is pathognomonic but is usually detectable only by a trained examiner. When this finding is present in the setting of suspected HPS, no preoperative imaging is necessary. Nonetheless, in most centers a pyloric sonogram will be obtained.
 The treatment is pyloromyotomy. Although spontaneous regression of muscular hypertrophy can occur, nonoperative therapy is difficult to justify given the fact that surgery is quick, safe, and effective.
The treatment is pyloromyotomy. Although spontaneous regression of muscular hypertrophy can occur, nonoperative therapy is difficult to justify given the fact that surgery is quick, safe, and effective.
Radiologic
 Plain film findings include (1) gastric distention, (2) mottled retained gastric contents, (3) indentations in the gastric wall by hyperperistaltic waves (i.e., so-called caterpillar stomach), and (4) diminished air beyond the pylorus.
Plain film findings include (1) gastric distention, (2) mottled retained gastric contents, (3) indentations in the gastric wall by hyperperistaltic waves (i.e., so-called caterpillar stomach), and (4) diminished air beyond the pylorus.
 Barium study findings include (1) delayed gastric emptying, (2) “string sign” through the pyloric channel, (3) double- or triple-track sign in pyloric channel, (4) “shouldering” in the antrum from muscular hypertrophy, (5) a “beak” sign of oral contrast material at the pyloric entrance, and (6) indentation on the duodenal bulb by the hypertrophied muscularis.
Barium study findings include (1) delayed gastric emptying, (2) “string sign” through the pyloric channel, (3) double- or triple-track sign in pyloric channel, (4) “shouldering” in the antrum from muscular hypertrophy, (5) a “beak” sign of oral contrast material at the pyloric entrance, and (6) indentation on the duodenal bulb by the hypertrophied muscularis.
 Sonographic findings include an elongated (i.e., ≥ 15 mm) pyloric channel and increased thickness (i.e., ≥ 3 mm) of the hypoechoic muscle, which are more sensitive findings for diagnosis of HPS than the cross-sectional diameter of the entire pylorus. A “target” or bull’s eye appearance of the pyloric channel is seen on transverse sonographic images.
Sonographic findings include an elongated (i.e., ≥ 15 mm) pyloric channel and increased thickness (i.e., ≥ 3 mm) of the hypoechoic muscle, which are more sensitive findings for diagnosis of HPS than the cross-sectional diameter of the entire pylorus. A “target” or bull’s eye appearance of the pyloric channel is seen on transverse sonographic images.
 Sonographic scanning at an angle tangential to the pylorus can cause a false-positive examination by producing “pseudothickening” of the muscularis. So-called pseudoelongation of the pyloric channel, another potential cause of false-positive examinations, can result from pylorospasm and use of prostaglandins.
Sonographic scanning at an angle tangential to the pylorus can cause a false-positive examination by producing “pseudothickening” of the muscularis. So-called pseudoelongation of the pyloric channel, another potential cause of false-positive examinations, can result from pylorospasm and use of prostaglandins.
SUGGESTED READING
Hernanz-Schulman M, Zhu Y, Stein SM, et al. Hypertrophic pyloric stenosis in infants: US evaluation of vascularity of the pyloric canal. Radiology 2003;229:389–393.
Hernanz-Schulman M. Infantile hypertrophic pyloric stenosis. Radiology 2003;22:319–331.
Hernanz-Schulman M, Sells L, Ambrosino MM, et al. Hypertrophic pyloric stenosis in the infant without a palpable olive: accuracy of sonographic diagnosis. Radiology 1994;193:771–776.
Liao Z, Li ZS, Zhang WJ, et al. Education and imaging. Gastrointestinal: infantile hypertrophic pyloric stenosis. J Gastroenterol Hepatol 2007;22:1692.
CAROLINE CARRICO
HISTORY
A newborn infant with torticollis and a palpable neck mass.
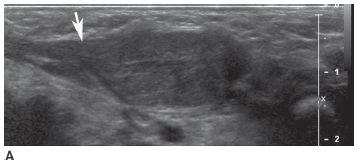
 FIGURE 8-7A Longitudinal sonographic image of the neck shows a solitary mass that is slightly heterogeneous and nearly isoechoic with the sternocleidomastoid muscle. The muscle has a fusiform appearance (arrow) caused by expansion of the muscle due to the fact that the lesion is contained within it.
FIGURE 8-7A Longitudinal sonographic image of the neck shows a solitary mass that is slightly heterogeneous and nearly isoechoic with the sternocleidomastoid muscle. The muscle has a fusiform appearance (arrow) caused by expansion of the muscle due to the fact that the lesion is contained within it.
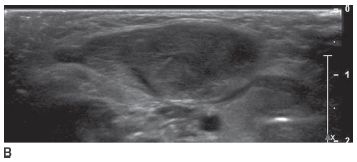
 FIGURE 8-7B Transverse sonographic image confirms that the lesion is isoechoic with muscle and is completely contained within the sternocleidomastoid muscle.
FIGURE 8-7B Transverse sonographic image confirms that the lesion is isoechoic with muscle and is completely contained within the sternocleidomastoid muscle.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis for an intramuscular mass in the sternocleidomastoid muscle in an infant with torticollis is very limited.
 Hemangiomas: These are common benign tumors of infancy that are typically dermal or subcutaneous. The classic lesions proliferate in the first few months of life and progressively become uniformly solid with a few associated high-flow vessels. They usually slowly involute over ensuing months or years. The purely intramuscular location and the lack of prominent central and peripheral vessels in the case shown here makes this consideration unlikely.
Hemangiomas: These are common benign tumors of infancy that are typically dermal or subcutaneous. The classic lesions proliferate in the first few months of life and progressively become uniformly solid with a few associated high-flow vessels. They usually slowly involute over ensuing months or years. The purely intramuscular location and the lack of prominent central and peripheral vessels in the case shown here makes this consideration unlikely.
 Infantile myofibromatosis: Infantile myofibromatosis is a very rare abnormality that affects children during their first two years of life. The lesions may be solitary or multicentric and typically present as a slow growing mass of fibrous tissue. Nearly one-third occur in the head and neck but they can also involve visceral organs (associated with a poorer prognosis). Although this diagnosis can not be absolutely excluded on the basis of the images shown in this case, the rarity of this entity makes it an unlikely diagnosis.
Infantile myofibromatosis: Infantile myofibromatosis is a very rare abnormality that affects children during their first two years of life. The lesions may be solitary or multicentric and typically present as a slow growing mass of fibrous tissue. Nearly one-third occur in the head and neck but they can also involve visceral organs (associated with a poorer prognosis). Although this diagnosis can not be absolutely excluded on the basis of the images shown in this case, the rarity of this entity makes it an unlikely diagnosis.
 Sarcoma: Three types of sarcoma could reasonably be considered in the case shown here.
Sarcoma: Three types of sarcoma could reasonably be considered in the case shown here.
 Rhabdomyosarcoma: Rhabdomyosarcoma can occur at any age and within any muscle. The margins of the lesion may be well-defined or poorly defined.
Rhabdomyosarcoma: Rhabdomyosarcoma can occur at any age and within any muscle. The margins of the lesion may be well-defined or poorly defined.
 Infantile fibrosarcoma: This is a childhood tumor that usually occurs in the legs, but can be found in any muscle. Both rhabdomyosarcoma and infantile fibrosarcoma could produce the imaging findings in the case shown here. However, they are both rare in the newborn period, making them unlikely diagnoses.
Infantile fibrosarcoma: This is a childhood tumor that usually occurs in the legs, but can be found in any muscle. Both rhabdomyosarcoma and infantile fibrosarcoma could produce the imaging findings in the case shown here. However, they are both rare in the newborn period, making them unlikely diagnoses.
 Granulocytic sarcoma (also known as myeloid sarcoma or chloroma): Granulocytic sarcoma can occur in any location and may be solitary or multiple. This rare tumor occurs in infants and children with myeloprolif-erative disorders such as acute or chronic myelogenous leukemia (which would be expected to produce anemia and easy brusiability, absent in the case presented here).
Granulocytic sarcoma (also known as myeloid sarcoma or chloroma): Granulocytic sarcoma can occur in any location and may be solitary or multiple. This rare tumor occurs in infants and children with myeloprolif-erative disorders such as acute or chronic myelogenous leukemia (which would be expected to produce anemia and easy brusiability, absent in the case presented here).
 Fibromatosis colli: This entity, also known as sterno-cleidomastoid pseudotumor of infancy or congenital torticollis, is a benign mass of fibrous tissue that arises within the sternocleidomastoid muscle during the first month of life. Most newborns with torticollis have fibromatosis colli. At sonography, the mass has variable echotexture and causes well-marginated fusiform enlargement of the affected sternocleidomastoid muscle (as is present in the images in the case shown here). This diagnosis is correct.
Fibromatosis colli: This entity, also known as sterno-cleidomastoid pseudotumor of infancy or congenital torticollis, is a benign mass of fibrous tissue that arises within the sternocleidomastoid muscle during the first month of life. Most newborns with torticollis have fibromatosis colli. At sonography, the mass has variable echotexture and causes well-marginated fusiform enlargement of the affected sternocleidomastoid muscle (as is present in the images in the case shown here). This diagnosis is correct.
 Lipoblastoma or lipoblastomatosis: As their names imply, these benign lesions contain fat as well as mysenchymal cells in a myxoid stroma. MR or CT imaging can confirm the presence of fat. Most lesions contain a large amount of fat, but some contain very little fat. The absence of fat makes this an unlikely consideration. Furthermore, lipoblastomastosis is more likely to extend to deeper tissues than in the case shown here.
Lipoblastoma or lipoblastomatosis: As their names imply, these benign lesions contain fat as well as mysenchymal cells in a myxoid stroma. MR or CT imaging can confirm the presence of fat. Most lesions contain a large amount of fat, but some contain very little fat. The absence of fat makes this an unlikely consideration. Furthermore, lipoblastomastosis is more likely to extend to deeper tissues than in the case shown here.
DIAGNOSIS
Fibromatosis colli
KEY FACTS
Clinical
 Fibromatosis colli is a nontender, benign mass within the sternocleidomastoid muscle of a neonate. The mass consists of fibroblasts, myoblasts, mesenchyme-like cells, and myofibroblasts.
Fibromatosis colli is a nontender, benign mass within the sternocleidomastoid muscle of a neonate. The mass consists of fibroblasts, myoblasts, mesenchyme-like cells, and myofibroblasts.
 Fibromatosis colli is a very common neck mass encountered in infancy; it is the most common noninflammatory mass.
Fibromatosis colli is a very common neck mass encountered in infancy; it is the most common noninflammatory mass.
 Most often the correct diagnosis is recognized clinically and no imaging is obtained.
Most often the correct diagnosis is recognized clinically and no imaging is obtained.
 The exact cause of these lesions is not known. The affected infant usually has a history of difficult or traumatic birth, for example, requiring forceps for delivery. Some infants have a history of abnormal positioning of the head in utero.
The exact cause of these lesions is not known. The affected infant usually has a history of difficult or traumatic birth, for example, requiring forceps for delivery. Some infants have a history of abnormal positioning of the head in utero.
 Twenty percent of affected neonates have torticollis with the head tilt and chin rotation toward the affected side. In the rare instance when lesions are bilateral, the neck appears short and the chin is tilted upward.
Twenty percent of affected neonates have torticollis with the head tilt and chin rotation toward the affected side. In the rare instance when lesions are bilateral, the neck appears short and the chin is tilted upward.
 Treatment is conservative. Physical therapy or no treatment may be used.
Treatment is conservative. Physical therapy or no treatment may be used.
 Spontaneous resolution usually occurs by 8 months of age.
Spontaneous resolution usually occurs by 8 months of age.
 It is uncommon for the abnormality to persist longer than a year or for facial asymmetry to develop. In these cases surgical intervention may be needed.
It is uncommon for the abnormality to persist longer than a year or for facial asymmetry to develop. In these cases surgical intervention may be needed.
Radiologic
 Sonography is often used to evaluate an infant with a nontraumatic palpable soft tissue abnormality in the neck.
Sonography is often used to evaluate an infant with a nontraumatic palpable soft tissue abnormality in the neck.
 At sonography, the mass of fibrous tissue within the sternocleidomastoid muscle caused by fibromatosis colli has variable echotexture, but is relatively homogeneous, and may be well-defined. The enlarged muscle is fusiform.
At sonography, the mass of fibrous tissue within the sternocleidomastoid muscle caused by fibromatosis colli has variable echotexture, but is relatively homogeneous, and may be well-defined. The enlarged muscle is fusiform.
 Imaging the sternocleidomastoid muscle in both the longitudinal axis and transverse axis is important to confirm that the lesion is completely intramuscular.
Imaging the sternocleidomastoid muscle in both the longitudinal axis and transverse axis is important to confirm that the lesion is completely intramuscular.
 Most cases are unilateral; thus, the uninvolved contralateral sternocleidomastoid muscle can be used for comparison.
Most cases are unilateral; thus, the uninvolved contralateral sternocleidomastoid muscle can be used for comparison.
 The affected portion of the sternocleidomastoid muscle may be hyperemic.
The affected portion of the sternocleidomastoid muscle may be hyperemic.
 If the diagnosis remains uncertain after sonography, MR imaging can show that the sternocleidomastoid muscle is enlarged and hyperintense or mildly hypoin-tense on T2-weighted images. However, in the case of fibromatosis colli, a focal mass may not be evident on MR imaging.
If the diagnosis remains uncertain after sonography, MR imaging can show that the sternocleidomastoid muscle is enlarged and hyperintense or mildly hypoin-tense on T2-weighted images. However, in the case of fibromatosis colli, a focal mass may not be evident on MR imaging.
 CT imaging of fibromatosis colli typically shows an enlarged sternocleidomastoid muscle with normal CT density.
CT imaging of fibromatosis colli typically shows an enlarged sternocleidomastoid muscle with normal CT density.
 Associated intramuscular hemorrhage or calcification has been occasionally described on CT and MR imaging.
Associated intramuscular hemorrhage or calcification has been occasionally described on CT and MR imaging.
SUGGESTED READING
Kumar V, Prabhu BV, Chattopadhayay A, Nagendhar MY. Bilateral sternocleidomastoid tumor of infancy. Int J Pediatr Otorhinolaryngol 2003;67:673–675.
Keller MS. Musculoskeletal sonography in the neonate and infant. Pediatr Radiol 2005;35:1167–1173; quiz 1293.
Laor T. MR imaging of soft tissue tumors and tumor-like lesions. Pediatr Radiol 2004;34:24–37.
Robbin MR, Murphey MD, Temple HT, et al. Imaging of musculoskeletal fibromatosis. Radiographics 2001;21:585–600.
DONALD P. FRUSH
HISTORY
A neonate with persistent right upper-lobe opacity on radiography.
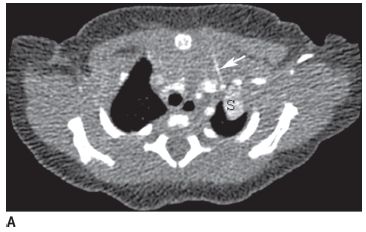
 FIGURE 8-8A Contrast-enhanced axial CT image shows left upper lobe mass (S) supplied by a small systemic artery (arrow) arising from the left internal mammary artery.
FIGURE 8-8A Contrast-enhanced axial CT image shows left upper lobe mass (S) supplied by a small systemic artery (arrow) arising from the left internal mammary artery.
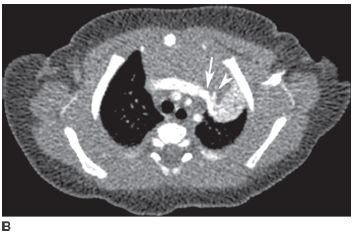
 FIGURE 8-8B Contrast-enhanced axial CT image at a more caudal level than shown in A depicts the distal course of the supplying artery (arrowhead). The venous drainage (arrow) is via the brachiocephalic vein.
FIGURE 8-8B Contrast-enhanced axial CT image at a more caudal level than shown in A depicts the distal course of the supplying artery (arrowhead). The venous drainage (arrow) is via the brachiocephalic vein.
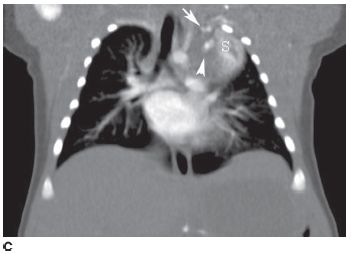
 FIGURE 8-8C Coronal image reconstructed from axial CT images shows the mass (S) is vertically oriented. The arterial supply (arrow) and venous drainage (arrowhead) are also shown.
FIGURE 8-8C Coronal image reconstructed from axial CT images shows the mass (S) is vertically oriented. The arterial supply (arrow) and venous drainage (arrowhead) are also shown.
 Atelectasis: This diagnosis is a reasonable consideration given the presence of the lung opacity. However, the vessels on the CT examination are not typical of atelectasis. Furthermore, atelectasis would not be expected to persist unchanged on serial radiographs.
Atelectasis: This diagnosis is a reasonable consideration given the presence of the lung opacity. However, the vessels on the CT examination are not typical of atelectasis. Furthermore, atelectasis would not be expected to persist unchanged on serial radiographs.
 Pulmonary sequestration: This diagnosis is the likeliest consideration (even though a lower lobe location is more typical), given that the opacity persisted. Furthermore, the arterial supply and venous drainage are characteristic of this entity.
Pulmonary sequestration: This diagnosis is the likeliest consideration (even though a lower lobe location is more typical), given that the opacity persisted. Furthermore, the arterial supply and venous drainage are characteristic of this entity.
 Bronchogenic cyst: This diagnosis is unlikely because a systemic vascular supply would not be expected.
Bronchogenic cyst: This diagnosis is unlikely because a systemic vascular supply would not be expected.
 Arteriovenous malformation (AVM): Pulmonary AVMs frequently have an appearance of multiple, mass-like lesions with prominent feeding arteries, which can be of systemic origin. However, in this case systemic (as opposed to pulmonary) venous drainage is present, which, by definition, is not possible with a pulmonary AVM. In addition, a mass of abnormal lung parenchyma would not be expected in an AVM, making this diagnosis unlikely.
Arteriovenous malformation (AVM): Pulmonary AVMs frequently have an appearance of multiple, mass-like lesions with prominent feeding arteries, which can be of systemic origin. However, in this case systemic (as opposed to pulmonary) venous drainage is present, which, by definition, is not possible with a pulmonary AVM. In addition, a mass of abnormal lung parenchyma would not be expected in an AVM, making this diagnosis unlikely.
 Pneumonia: The clinical history is not consistent with pneumonia. Furthermore, the prominent vascularity associated with this lesion excludes acute pneumonia.
Pneumonia: The clinical history is not consistent with pneumonia. Furthermore, the prominent vascularity associated with this lesion excludes acute pneumonia.
 Pulmonary neoplasm: Because pulmonary neoplasms are very rare lesions in neonates and because blood supply from the aorta would not be expected in a neoplasm, this diagnosis is very unlikely.
Pulmonary neoplasm: Because pulmonary neoplasms are very rare lesions in neonates and because blood supply from the aorta would not be expected in a neoplasm, this diagnosis is very unlikely.
 Chest wall mass: The CT in this case indicates that the mass is within the lung parenchyma as opposed to the chest wall. Furthermore, chest wall masses are rare in neonates (and much less common than pulmonary sequestration). Finally, a chest wall mass would not be expected to have the large vessel vascular pattern that is present in the case illustrated here. This diagnosis is incorrect.
Chest wall mass: The CT in this case indicates that the mass is within the lung parenchyma as opposed to the chest wall. Furthermore, chest wall masses are rare in neonates (and much less common than pulmonary sequestration). Finally, a chest wall mass would not be expected to have the large vessel vascular pattern that is present in the case illustrated here. This diagnosis is incorrect.
DIAGNOSIS
Pulmonary sequestration (intralobar type)
KEY FACTS
Clinical
 The term sequestration refers to a nonfunctioning pulmonary parenchymal abnormality that lacks normal tracheobronchial communication and has anomalous systemic (rather than pulmonary) arterial supply. Venous drainage may be via systemic or pulmonary veins.
The term sequestration refers to a nonfunctioning pulmonary parenchymal abnormality that lacks normal tracheobronchial communication and has anomalous systemic (rather than pulmonary) arterial supply. Venous drainage may be via systemic or pulmonary veins.
 Seuestration can occur with other congenital lung abnormalities (e.g., bronchogenic cyst, congenital cystic adenomatoid malformation, congenital lobar emphysema [CLE]), referred to as a hybrid abnormality.
Seuestration can occur with other congenital lung abnormalities (e.g., bronchogenic cyst, congenital cystic adenomatoid malformation, congenital lobar emphysema [CLE]), referred to as a hybrid abnormality.
 Sequestration is part of a continuum of bronchopul-monary foregut malformations (which have variable contributions of anomalies of lung, airway, and vascu-lature). Traditionally, a distinction is made between an intralobar (i.e., no separate pleura) and an extralobar (i.e., having a separate pleural investment) type of sequestration.
Sequestration is part of a continuum of bronchopul-monary foregut malformations (which have variable contributions of anomalies of lung, airway, and vascu-lature). Traditionally, a distinction is made between an intralobar (i.e., no separate pleura) and an extralobar (i.e., having a separate pleural investment) type of sequestration.
 Modes of presentation generally differ according to the age at the time of diagnosis.
Modes of presentation generally differ according to the age at the time of diagnosis.
 In the prenatal period, the diagnosis is based on the finding of an echogenic chest mass at sonography or fetal MRI.
In the prenatal period, the diagnosis is based on the finding of an echogenic chest mass at sonography or fetal MRI.
 The clinical presentation in the neonatal period can be respiratory distress. However, infants can be asymp-tomic.
The clinical presentation in the neonatal period can be respiratory distress. However, infants can be asymp-tomic.
 In a child (and occasionally an adult), the clinical presentation is that of recurrent pulmonary infections, with the sequestration often initially being misdiag-nosed as pneumonia. The sequestration is most often an “incidental” finding unrelated to clinical findings, suggesting lower respiratory infection.
In a child (and occasionally an adult), the clinical presentation is that of recurrent pulmonary infections, with the sequestration often initially being misdiag-nosed as pneumonia. The sequestration is most often an “incidental” finding unrelated to clinical findings, suggesting lower respiratory infection.
 More than half of sequestrations present in childhood. Most extralobar sequestrations are discovered within the first year of life.
More than half of sequestrations present in childhood. Most extralobar sequestrations are discovered within the first year of life.
 Sequestration can occasionally present in an infant or small child as a murmur with congestive heart failure due to the arteriovenous shunting.
Sequestration can occasionally present in an infant or small child as a murmur with congestive heart failure due to the arteriovenous shunting.
 The intralobar type is more common and can be congenital or acquired. Most are lower lobe in location, with two-thirds located on the left side. Drainage is usually via the pulmonary veins.
The intralobar type is more common and can be congenital or acquired. Most are lower lobe in location, with two-thirds located on the left side. Drainage is usually via the pulmonary veins.
 The extralobar type also usually occurs in the lower lobe and is on the left side in 90% of cases. This type may also be subdiaphragmatic. Drainage is typically through systemic veins. Fifty percent of cases have associated anomalies e.g., heart disease, diaphragmatic hernias, gastrointestinal, and other pulmonary abnormalities.
The extralobar type also usually occurs in the lower lobe and is on the left side in 90% of cases. This type may also be subdiaphragmatic. Drainage is typically through systemic veins. Fifty percent of cases have associated anomalies e.g., heart disease, diaphragmatic hernias, gastrointestinal, and other pulmonary abnormalities.
 Treatment for pulmonary sequestrations is surgical excision, which may be deferred for a period of 6 to 12 months, if the neonate is asymptomatic.
Treatment for pulmonary sequestrations is surgical excision, which may be deferred for a period of 6 to 12 months, if the neonate is asymptomatic.
Radiologic
 The sequestered lung can receive air flow through collateral sources. Therefore, if recurrent infections have not occurred, the radiographic appearance can be normal.
The sequestered lung can receive air flow through collateral sources. Therefore, if recurrent infections have not occurred, the radiographic appearance can be normal.
 The diagnosis should be considered when persistent or recurrent lower lobe opacity is found.
The diagnosis should be considered when persistent or recurrent lower lobe opacity is found.
 Catheter angiography has been replaced by other, noninvasive imaging studies to determine vascular supply. Multidetector computed tomography (MDCT) angiogra-phy is the preferred method of diagnosis but MRI can also be especially useful.
Catheter angiography has been replaced by other, noninvasive imaging studies to determine vascular supply. Multidetector computed tomography (MDCT) angiogra-phy is the preferred method of diagnosis but MRI can also be especially useful.
 Sonography with color Doppler can be used to document arterial supply if a good acoustic window is present to facilitate imaging; however, usually cross-sectional imaging is performed prior to complete evaluation.
Sonography with color Doppler can be used to document arterial supply if a good acoustic window is present to facilitate imaging; however, usually cross-sectional imaging is performed prior to complete evaluation.
SUGGESTED READING
Frush DP, Donnelly LE. Pulmonary sequestration: a new spin with helical CT. AJR Am J Roentgenol 1997;169:679–682.
Kocaoglu M, Frush DP, Ugurel MS, Somuncu I. Bronchopulmonary fore-gut malformations presenting as mass lesions in children: spectrum of imaging findings. Diagn Interv Radiol 2010;16:153–161.
Newman B. Congenital bronchopulmonary foregut malformations: concepts and controversies. Pediatr Radiol 2006;36:773–791.
ANA M. GACA
HISTORY
A three-week-old girl with a swollen left leg and foot who refuses to move her left leg. Her mother reports falling down stairs while holding the infant the day before imaging was performed.
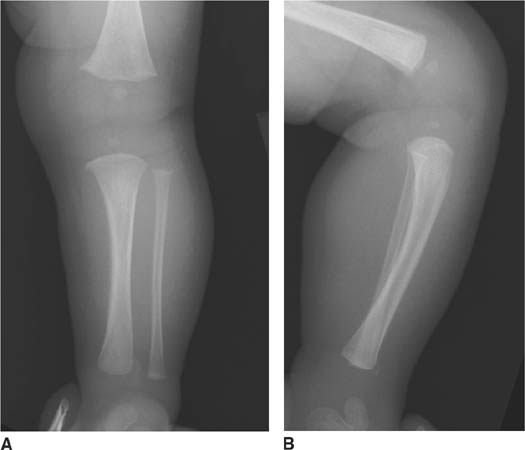
 FIGURE 8-9A and 8-9B Left lower extremity radiograph. A classic metaphyseal lesion (CML), also known as a corner fracture or a bucket-handle fracture (different projections of the same fracture), is present at the anterior aspect of the distal tibial metaphysis.
FIGURE 8-9A and 8-9B Left lower extremity radiograph. A classic metaphyseal lesion (CML), also known as a corner fracture or a bucket-handle fracture (different projections of the same fracture), is present at the anterior aspect of the distal tibial metaphysis.
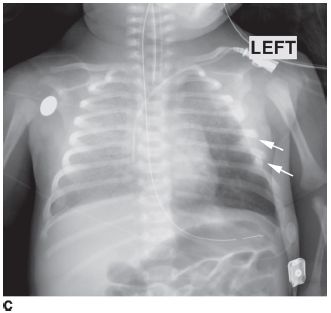
 FIGURE 8-9C Chest radiograph on a different patient, a 1-month-old girl who presented with respiratory difficulty. Multiple acute left lateral rib fracture (arrows) and bilateral pulmonary opacities, worse in the right lung, are present.
FIGURE 8-9C Chest radiograph on a different patient, a 1-month-old girl who presented with respiratory difficulty. Multiple acute left lateral rib fracture (arrows) and bilateral pulmonary opacities, worse in the right lung, are present.
DIFFERENTIAL DIAGNOSIS
 Nonaccidental trauma (infant/child abuse): This diagnosis is a likely consideration given the type of fracture, the classic metaphyseal lesion (CML) seen in nonaccidental trauma. In the case presented here, the pulmonary opacities are further suggestive of nonaccidental trauma because they are consistent with pulmonary contusion. In combination with the rib fractures and other fractures, nonaccidental trauma is the most likely diagnosis. As discussed below, the clinical information in any individual case is important to rule out other entities that can mimic nonaccidental trauma.
Nonaccidental trauma (infant/child abuse): This diagnosis is a likely consideration given the type of fracture, the classic metaphyseal lesion (CML) seen in nonaccidental trauma. In the case presented here, the pulmonary opacities are further suggestive of nonaccidental trauma because they are consistent with pulmonary contusion. In combination with the rib fractures and other fractures, nonaccidental trauma is the most likely diagnosis. As discussed below, the clinical information in any individual case is important to rule out other entities that can mimic nonaccidental trauma.
 Accidental trauma: A CML, also known as a “corner” fracture, would not be expected to occur from accidental trauma. Fractures from accidental trauma are typically spiral or oblique fractures (often in the diaphysis) or so-called buckle fractures (commonly of the metaph-ysis or metadiaphysis). This diagnosis is incorrect.
Accidental trauma: A CML, also known as a “corner” fracture, would not be expected to occur from accidental trauma. Fractures from accidental trauma are typically spiral or oblique fractures (often in the diaphysis) or so-called buckle fractures (commonly of the metaph-ysis or metadiaphysis). This diagnosis is incorrect.
 Osteogenesis imperfecta (OI): The presence of multiple fractures raises this entity as a possible diagnosis. Clinical findings associated with OI include multiple fractures and bony deformity, blue sclera, hearing loss, tooth abnormalities (dentinogenesis imperfecta), and joint laxity, which are not reported to be present in this case.
Osteogenesis imperfecta (OI): The presence of multiple fractures raises this entity as a possible diagnosis. Clinical findings associated with OI include multiple fractures and bony deformity, blue sclera, hearing loss, tooth abnormalities (dentinogenesis imperfecta), and joint laxity, which are not reported to be present in this case.
 Menkes’ syndrome (copper malabsorption): The clinical history and physical examination are especially important for exclusion of this diagnosis. Patients with this very rare syndrome may have failure to thrive, delayed developmental milestones, and kinky hair, eyebrows, and eyelashes. They later develop mental retardation and typically die in infancy. Such features are not present in the case presented, excluding the diagnosis.
Menkes’ syndrome (copper malabsorption): The clinical history and physical examination are especially important for exclusion of this diagnosis. Patients with this very rare syndrome may have failure to thrive, delayed developmental milestones, and kinky hair, eyebrows, and eyelashes. They later develop mental retardation and typically die in infancy. Such features are not present in the case presented, excluding the diagnosis.
 Metaphyseal irregularity as a normal variant: This variant is seen at the metaphyses as a beak or spur of bone that is continuous with the cortical bone. However, in the case presented here, the spur is not continuous, indicating that a fracture (i.e., a CML) is present. In questionable cases, a follow-up skeletal survey intended to identify other fractures can help distinguish a CML from a normal variant. Metaphyseal irreglarity is an incorrect diagnosis.
Metaphyseal irregularity as a normal variant: This variant is seen at the metaphyses as a beak or spur of bone that is continuous with the cortical bone. However, in the case presented here, the spur is not continuous, indicating that a fracture (i.e., a CML) is present. In questionable cases, a follow-up skeletal survey intended to identify other fractures can help distinguish a CML from a normal variant. Metaphyseal irreglarity is an incorrect diagnosis.
 Rickets: Congenital rickets due to maternal vitamin D deficiency can predispose a neonate or infant to insufficiency fractures. Diffuse osteopenia is often present; multiple fractures may also be encountered. The lack of oseteopenia in the case presented here makes the diagnosis of rickets unlikely.
Rickets: Congenital rickets due to maternal vitamin D deficiency can predispose a neonate or infant to insufficiency fractures. Diffuse osteopenia is often present; multiple fractures may also be encountered. The lack of oseteopenia in the case presented here makes the diagnosis of rickets unlikely.
DIAGNOSIS
Nonaccidental trauma (i.e., child abuse)
KEY FACTS
Clinical
 In addition to the multiple types of skeletal injuries seen in the setting of abuse, central nervous system injury (e.g., subdural hematoma, diffuse edema, diffuse infarction from hypoxia, and parenchymal hemorrhage) may be present. The mechanism for these injuries is currently thought to be impact against a soft object in combination with shaking, rather than (as previously thought) shaking alone.
In addition to the multiple types of skeletal injuries seen in the setting of abuse, central nervous system injury (e.g., subdural hematoma, diffuse edema, diffuse infarction from hypoxia, and parenchymal hemorrhage) may be present. The mechanism for these injuries is currently thought to be impact against a soft object in combination with shaking, rather than (as previously thought) shaking alone.
 Rib fractures are more common in children younger than 1 year old, whereas diaphyseal injuries are more common in those older than 1 year old. This fact reflects the manner in which the child is typically held during the trauma.
Rib fractures are more common in children younger than 1 year old, whereas diaphyseal injuries are more common in those older than 1 year old. This fact reflects the manner in which the child is typically held during the trauma.
 Direct blows to the abdomen most frequently result in duodenal hematoma, pancreatitis (with its attendant complications), and hepatic and splenic injuries.
Direct blows to the abdomen most frequently result in duodenal hematoma, pancreatitis (with its attendant complications), and hepatic and splenic injuries.
 The diagnosis of nonaccidental trauma is established using a combination of history (i.e., incompatibility of the reported history in relation to the type and extent of injuries), physical findings (e.g., burns, hand marks, sexual abuse, preretinal hemorrhages), and radiographic findings.
The diagnosis of nonaccidental trauma is established using a combination of history (i.e., incompatibility of the reported history in relation to the type and extent of injuries), physical findings (e.g., burns, hand marks, sexual abuse, preretinal hemorrhages), and radiographic findings.
 A major diagnostic consideration is OI, in which a family history of fractures, deafness, blue sclerae, and poor dentition may be elicited. If necessary, the diagnosis can be confirmed with a skin biopsy demonstrating a collagen synthesis deficiency.
A major diagnostic consideration is OI, in which a family history of fractures, deafness, blue sclerae, and poor dentition may be elicited. If necessary, the diagnosis can be confirmed with a skin biopsy demonstrating a collagen synthesis deficiency.
 Another diagnostic consideration is Menkes’ syndrome, a very rare neurodegenerative disorder due to a defect in copper metabolism, leading to death within the first year of life. These patients have sparse, brittle hair, which accounts for the designation “kinky hair syndrome.”
Another diagnostic consideration is Menkes’ syndrome, a very rare neurodegenerative disorder due to a defect in copper metabolism, leading to death within the first year of life. These patients have sparse, brittle hair, which accounts for the designation “kinky hair syndrome.”
 Congenital rickets due to maternal vitamin D deficiency is a consideration in cases of suspected nonaccidental trauma, although there is still active debate over the degree of overlap in terms of clinical features between the two disease states.
Congenital rickets due to maternal vitamin D deficiency is a consideration in cases of suspected nonaccidental trauma, although there is still active debate over the degree of overlap in terms of clinical features between the two disease states.
Radiologic
 Child or infant abuse can have central nervous system, abdominal, or skeletal findings evident on imaging studies. The skeletal manifestations are reviewed below.
Child or infant abuse can have central nervous system, abdominal, or skeletal findings evident on imaging studies. The skeletal manifestations are reviewed below.
 The CML is the most specific injury in child abuse. The CML is seen almost exclusively in children younger than 2 years old and most commonly seen in the distal femur, proximal tibia, distal tibia and proximal humerus.
The CML is the most specific injury in child abuse. The CML is seen almost exclusively in children younger than 2 years old and most commonly seen in the distal femur, proximal tibia, distal tibia and proximal humerus.
 Depending on the radiographic projection, a CML may appear as a corner fracture or a so-called bucket handle fracture.
Depending on the radiographic projection, a CML may appear as a corner fracture or a so-called bucket handle fracture.
 In addition to the CML, other injuries that are highly specific for child abuse include fractures in unusual locations such as the scapulae, sternum, and posterior ribs.
In addition to the CML, other injuries that are highly specific for child abuse include fractures in unusual locations such as the scapulae, sternum, and posterior ribs.
 Injuries that are moderately specific for child abuse include fractures of varying ages, complex skull fractures, and fractures involving the digits or vertebral bodies.
Injuries that are moderately specific for child abuse include fractures of varying ages, complex skull fractures, and fractures involving the digits or vertebral bodies.
 Spiral fractures are injuries with low specificity for child abuse. However, suspicion should be raised when such fractures are seen in children who are not old enough to have engaged in activities that would make an accidental cause likely.
Spiral fractures are injuries with low specificity for child abuse. However, suspicion should be raised when such fractures are seen in children who are not old enough to have engaged in activities that would make an accidental cause likely.
 One spiral fracture that should not raise suspicion for child abuse is the “toddler’s fracture,” a spiral fracture of the distal tibia. This injury is seen in children who are learning to walk and is not an indication for performing a skeletal survey.
One spiral fracture that should not raise suspicion for child abuse is the “toddler’s fracture,” a spiral fracture of the distal tibia. This injury is seen in children who are learning to walk and is not an indication for performing a skeletal survey.
 Physiologic periostitis in the infant is a normal bony condition, which is typically seen between 1 and 6 months of age. It is characteristically manifested as a single (or occasionally multiple) thin layer of periosteal reaction on the medial aspect of the femur, tibia, humerus, and radius, and is symmetric in distribution.
Physiologic periostitis in the infant is a normal bony condition, which is typically seen between 1 and 6 months of age. It is characteristically manifested as a single (or occasionally multiple) thin layer of periosteal reaction on the medial aspect of the femur, tibia, humerus, and radius, and is symmetric in distribution.
 The radionuclide bone scan (and potentially whole-body MR imaging) is a complementary method to plain films for detection of injuries due to abuse. This imaging study is particularly sensitive for detection of posterior rib fractures as well as diaphyseal injuries (which may actually be bowing fractures that can be inapparent on plain films).
The radionuclide bone scan (and potentially whole-body MR imaging) is a complementary method to plain films for detection of injuries due to abuse. This imaging study is particularly sensitive for detection of posterior rib fractures as well as diaphyseal injuries (which may actually be bowing fractures that can be inapparent on plain films).
SUGGESTED READING
Kleinman PK. Diagnostic Imaging in Infant Abuse. AJR Am J Roentgenol 1990;155:703–712.
Lonergan GJ, Baker MB, Morey MK, Boos SC. From the Archives of the AFIP. Child abuse: radiologic-pathologic correlation. Radiographs 2003;23:811–845.
Nimkin K, Kleinman PK. Imaging of child abuse. Radiol Clin North Am 2001;39:843–864.
Slovis TL, Chapman S. Evaluating the data concerning vitamin D insuf-flciency/deflciency and child abuse. Pediatr Radiol 2008;38:1221–1224.
CHARLES MAXFIELD
HISTORY
A neonate with low hematocrit following traumatic delivery.
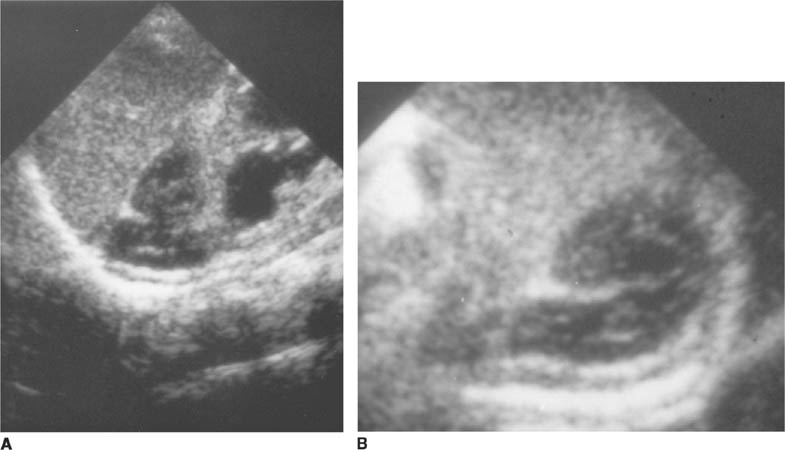
 FIGURES 8-10A and 8-10B Longitudinal (A) and transverse (B) views from a left upper quadrant sonogram. A structure with heterogeneous echogenicity is seen just superior to the left kidney in the expected location of the left adrenal gland. The structure has the pyramidal shape expected of the adrenal gland (although the limbs are enlarged). Dilatation of the renal collecting system is seen but is an incidental finding. The right adrenal gland (not shown) had a normal appearance.
FIGURES 8-10A and 8-10B Longitudinal (A) and transverse (B) views from a left upper quadrant sonogram. A structure with heterogeneous echogenicity is seen just superior to the left kidney in the expected location of the left adrenal gland. The structure has the pyramidal shape expected of the adrenal gland (although the limbs are enlarged). Dilatation of the renal collecting system is seen but is an incidental finding. The right adrenal gland (not shown) had a normal appearance.
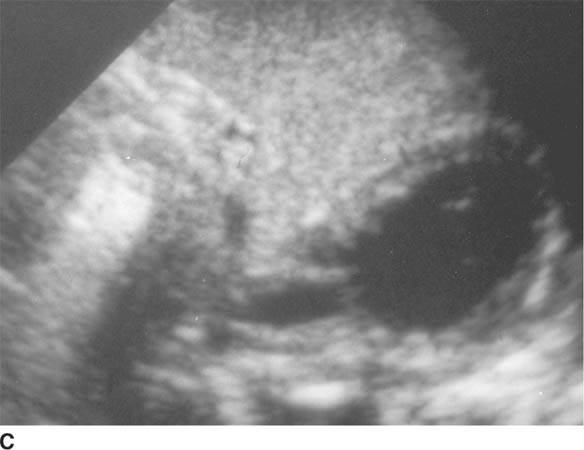
 FIGURE 8-10C Transverse sonogram 10 days after Figures 8-10A and 8-10B were obtained. During the period since the initial study, the lesion has become hypoechoic, with an anechoic central region. A slightly irregular echogenic rim is evident.
FIGURE 8-10C Transverse sonogram 10 days after Figures 8-10A and 8-10B were obtained. During the period since the initial study, the lesion has become hypoechoic, with an anechoic central region. A slightly irregular echogenic rim is evident.
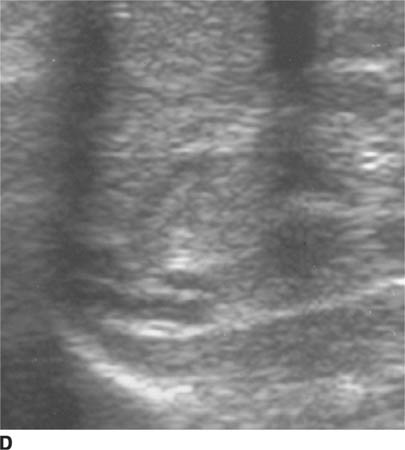
 FIGURE 8-10D Transverse sonogram of adrenal gland in a different patient. An adrenal gland in a normal neonate is illustrated for comparison to the previous images. The gland has symmetric, hypoechoic limbs with a central echogenic stripe.
FIGURE 8-10D Transverse sonogram of adrenal gland in a different patient. An adrenal gland in a normal neonate is illustrated for comparison to the previous images. The gland has symmetric, hypoechoic limbs with a central echogenic stripe.
 Congenital neuroblastoma: This entity typically has the radiologic appearance of a solid (rather than cystic) mass, whereas the lesions shown in the case presented here are hypoechoic (consistent with fluid). Although neuroblastoma could become cystic following chemotherapy or a biopsy producing internal hemorrhage, no history of these events is present in the patient shown here. Finally, neuroblastoma tends to distort the shape of the adrenal gland; however, in the case presented, the shape of the adrenal gland is preserved. For these reasons, neuroblastoma is an unlikely consideration.
Congenital neuroblastoma: This entity typically has the radiologic appearance of a solid (rather than cystic) mass, whereas the lesions shown in the case presented here are hypoechoic (consistent with fluid). Although neuroblastoma could become cystic following chemotherapy or a biopsy producing internal hemorrhage, no history of these events is present in the patient shown here. Finally, neuroblastoma tends to distort the shape of the adrenal gland; however, in the case presented, the shape of the adrenal gland is preserved. For these reasons, neuroblastoma is an unlikely consideration.
 Other retroperitoneal tumors (e.g., lymphangioma, teratoma): These tumors are very rarely located in the retroperitoneum. Furthermore, such lesions are usually calcified, fluid-filled masses. This diagnosis is an unlikely one.
Other retroperitoneal tumors (e.g., lymphangioma, teratoma): These tumors are very rarely located in the retroperitoneum. Furthermore, such lesions are usually calcified, fluid-filled masses. This diagnosis is an unlikely one.
 Perinephric abscess: The mixed echogenicity seen in the case presented here could be seen in a phlegmon or abscess. However, no history of infection is present; furthermore, such a process would be very unusual in a newborn. Finally, a perinephric abscess would be expected to arise adjacent to, rather than within, the adrenal gland). However, in this case shown, the adrenal gland itself is the site of the abnormality. Perineph-ric abscess is an incorrect diagnosis.
Perinephric abscess: The mixed echogenicity seen in the case presented here could be seen in a phlegmon or abscess. However, no history of infection is present; furthermore, such a process would be very unusual in a newborn. Finally, a perinephric abscess would be expected to arise adjacent to, rather than within, the adrenal gland). However, in this case shown, the adrenal gland itself is the site of the abnormality. Perineph-ric abscess is an incorrect diagnosis.
 Congenital adrenal hyperplasia: This entity might be considered because of the enlargement of the adrenal glands. In congenital adrenal hyperplasia, the glands do maintain their adreniform shape. However, enlargement of both glands is present, whereas, in the case shown here, a single adrenal gland is affected. Endo-crinologic abnormalities are helpful in supporting the diagnosis of congenital adrenal hyperplasia. However, we are given no history of such abnormalities in the case presented here. Therefore, this diagnosis is unlikely.
Congenital adrenal hyperplasia: This entity might be considered because of the enlargement of the adrenal glands. In congenital adrenal hyperplasia, the glands do maintain their adreniform shape. However, enlargement of both glands is present, whereas, in the case shown here, a single adrenal gland is affected. Endo-crinologic abnormalities are helpful in supporting the diagnosis of congenital adrenal hyperplasia. However, we are given no history of such abnormalities in the case presented here. Therefore, this diagnosis is unlikely.
 Adrenal hemorrhage: In the case shown, the echotex-ture of the mass changed relatively quickly, which would be unusual for any entity other than hemorrhage. Furthermore, the shape of the adrenal gland is maintained, which would be unusual for some of the diagnostic considerations other than hemorrhage but common for adrenal hemorrhage. This diagnosis is correct.
Adrenal hemorrhage: In the case shown, the echotex-ture of the mass changed relatively quickly, which would be unusual for any entity other than hemorrhage. Furthermore, the shape of the adrenal gland is maintained, which would be unusual for some of the diagnostic considerations other than hemorrhage but common for adrenal hemorrhage. This diagnosis is correct.
DIAGNOSIS
Neonatal adrenal hemorrhage
KEY FACTS
Clinical
 Spontaneous adrenal hemorrhage occurs relatively commonly in newborn infants and is occasionally detected in utero.
Spontaneous adrenal hemorrhage occurs relatively commonly in newborn infants and is occasionally detected in utero.
 Predisposing factors include perinatal stress, traumatic birth, hypoxia, and sepsis. Hemorrhage also occurs with increased frequency in large infants and infants of diabetic mothers.
Predisposing factors include perinatal stress, traumatic birth, hypoxia, and sepsis. Hemorrhage also occurs with increased frequency in large infants and infants of diabetic mothers.
 Large hemorrhages can clinically present with systemic shock or a palpable mass; smaller hemorrhages cause mild anemia or jaundice. Biochemical abnormalities are seldom present in either the acute or convalescent phase.
Large hemorrhages can clinically present with systemic shock or a palpable mass; smaller hemorrhages cause mild anemia or jaundice. Biochemical abnormalities are seldom present in either the acute or convalescent phase.
 Hemorrhage is more common in the left adrenal gland and is seen bilaterally in approximately 10% of cases.
Hemorrhage is more common in the left adrenal gland and is seen bilaterally in approximately 10% of cases.
 Complications are uncommon but include renal vein thrombosis (especially when the left adrenal gland is involved) and secondary infection/abscess.
Complications are uncommon but include renal vein thrombosis (especially when the left adrenal gland is involved) and secondary infection/abscess.
Radiologic
 The primary diagnostic considerations for a suprarenal mass in a neonate are acute adrenal hemorrhage and congenital neuroblastoma.
The primary diagnostic considerations for a suprarenal mass in a neonate are acute adrenal hemorrhage and congenital neuroblastoma.
 Doppler evaluation can help distinguish adrenal hemorrhage from neuroblastoma based on vascularity; a hematoma is typically avascular while a tumor is generally well vascularized.
Doppler evaluation can help distinguish adrenal hemorrhage from neuroblastoma based on vascularity; a hematoma is typically avascular while a tumor is generally well vascularized.
 At sonography, hemorrhage is typically initially echo-genic, next develops mixed echogenicity as the clot retracts, and becomes hypo- or anechoic as it liquefies.
At sonography, hemorrhage is typically initially echo-genic, next develops mixed echogenicity as the clot retracts, and becomes hypo- or anechoic as it liquefies.
 Serial sonographic studies documenting rapidly decreasing size of the mass are consistent with adrenal hemorrhage. Progressive enlargement of the mass suggests a diagnosis of neuroblastoma and is an indication for surgical resection.
Serial sonographic studies documenting rapidly decreasing size of the mass are consistent with adrenal hemorrhage. Progressive enlargement of the mass suggests a diagnosis of neuroblastoma and is an indication for surgical resection.
 MR imaging has been used to establish the diagnosis but often does not definitively distinguish adrenal hemorrhage from neuroblastoma. Fortunately, small neu-roblastomas in the newborn invariably have favorable histology, which allows serial imaging with sonography without substantial risk of rapid growth.
MR imaging has been used to establish the diagnosis but often does not definitively distinguish adrenal hemorrhage from neuroblastoma. Fortunately, small neu-roblastomas in the newborn invariably have favorable histology, which allows serial imaging with sonography without substantial risk of rapid growth.
 Dystrophic calcifications can be the first radiologic indication of previous adrenal hemorrhage. Adrenal calcification, however, can also be seen in many other entities, such as neuroblastoma, ganglioneuroblastoma, Wolman’s syndrome, pheochromocytoma, tuberculosis, and adrenal carcinoma.
Dystrophic calcifications can be the first radiologic indication of previous adrenal hemorrhage. Adrenal calcification, however, can also be seen in many other entities, such as neuroblastoma, ganglioneuroblastoma, Wolman’s syndrome, pheochromocytoma, tuberculosis, and adrenal carcinoma.
SUGGESTED READING
Deeg KH, Bettendorf U, Hofmann V. Differential diagnosis of neonatal adrenal haemorrhage and congenital neuroblastoma by colour coded Doppler sonography and power Doppler sonography. Eur J Pediatr 1998;157:294–297.
Friedman GK, Castleberry RP. Changing trends of research and treatment in infant neuroblastoma. Pediatr Blood Cancer 2007;49:1060–1065.
Hendry GMA. Cystic neuroblastoma of the adrenal gland- A potential source of error in ultrasonic diagnosis. Pediatr Radiol 1982;12:204–206.
Nuchtern JG. Perinatal neuroblastoma. Semin Pediatr Surg 2006;15:10–16.
Velaphi SC, Perlman JM. Neonatal adrenal hemorrhage: clinical and abdominal sonographic findings. Clin Pediatr (Phila) 2001;40:545–548.
Westra SJ, Zaninovic AC, Hall TR, et al. Imaging of the adrenal gland in children. Radiographics 1994;14:1323–1340.
DONALD P. FRUSH
HISTORY
A term neonate with mild tachypnea but no cyanosis.
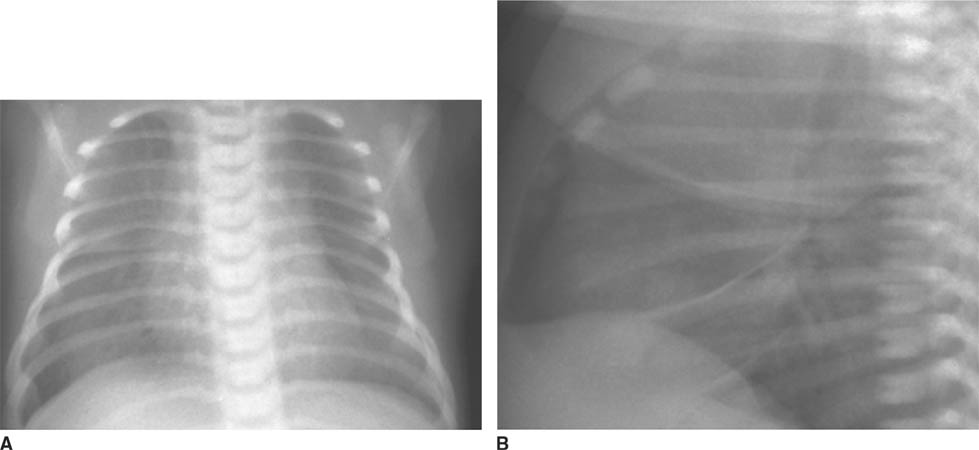
 FIGURES 8-11A and 8-11B Frontal (A) and lateral (B) chest radiograph (2 hours postnatal). Normal lung volumes are present, with streaky hilar opacities, indistinct vascularity, and subpleural and pleural fluid. The heart size is normal.
FIGURES 8-11A and 8-11B Frontal (A) and lateral (B) chest radiograph (2 hours postnatal). Normal lung volumes are present, with streaky hilar opacities, indistinct vascularity, and subpleural and pleural fluid. The heart size is normal.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


