Fig. 14.1
Neurosteroidogenesis: Steroidogenesis follows a sequential, highly compartmentalized reaction, with the translocation of cholesterol from cytoplasm to mitochondria in the cells of CNS, which is mediated by steroidogenic acute regulatory protein (StAR) and an 18 kDa translocator protein (TSPO). In the mitochondria, P450 side-chain cleavage (P450SCC) cleaves the side chain from cholesterol, resulting in the formation of pregnenolone. Pregnenolone is subsequently converted to progesterone and dehydroepiandrosterone in endoplasmic reticulum. The lipophilic nature of these compounds allows them to diffuse from one cell to another. Progesterone and dehydroepiandrosterone are further metabolized to form other neuroactive metabolites like testosterone, estrone, and estradiol
In the central nervous system (CNS), neuroactive steroids/neurosteroids play a crucial role in neuronal development and plasticity. Neuroactive steroids can act as allosteric modulators of ligand-gated ion channels (GABAA), NMDA, and sigma receptors. Because of these interactions, neuroactive steroids have been implicated to have sleep-inducing, anticonvulsant, anesthetic, nootropic, and antipsychotic properties (Rupprecht 2003). They are also associated with processes like learning, memory, emotion, behavior, synaptic transmission, and neuroprotection (Giatti et al. 2012). Because of these neuroprotective properties, steroid hormones may have a beneficial effect in neurodegenerative diseases, including Alzheimer’s disease, multiple sclerosis, Parkinson’s disease, and Huntington’s disease. However, steroid hormones are also implicated in psychiatric disorders, due to their role in cognition and behavior.
14.2 Steroid Hormones in Brain Disorders
14.2.1 Estrogens
Women have a higher prevalence of reduced cognitive function, depression, panic disorder, generalized anxiety disorder, social phobia, eating disorders, and some personality traits than men (Holsen et al. 2011). This difference in prevalence between sexes suggests that female sex steroid hormones may be involved in these conditions. This hypothesis is supported by the observation that several mental changes occur during the transition of women from pre- to postmenopause. These changes include reduced sexual drive, premenstrual and perimenopausal dysphoria induced by oral contraceptives or hormone replacement therapy (Eriksson et al. 2002; Rubinow and Schmidt 2006; Steiner et al. 2003). These symptoms are associated with altered levels of circulating estrogens. At menopause, levels of circulating estrogens are strongly reduced. This reduction in estrogen levels is associated with social and psychological changes in women, like anxiety, irritability, stress, memory loss, lack of concentration, and loss of libido. Eventually, this may culminate into depression (Bryant et al. 2012). The effects of estrogen changes are likely the result of an altered interaction of estrogens with other neurotransmitters like acetylcholine, dopamine, noradrenaline, and serotonin (McEwen 2001). Moreover, prolonged deficiency of estrogens increases the risk to develop dementia (Sherman et al. 2005) and Alzheimer’s disease (Pike et al. 2009).
As a consequence, maintenance of steady levels of estrogens seems essential for normal physiological and mental status. In fact, estrogen replacement therapy was found to improve learning, memory, and cognition in postmenopausal women (Sherwin 1997) and to prevent depression both in peri- and postmenopausal women (Grigoriadis and Kennedy 2002). Furthermore, estrogen replacement therapy was also found to have a protective effect against neurodegenerative disorders. This protective effect was ascribed to the anti-inflammatory activity of estrogens (Resnick and Henderson 2002; Vegeto et al. 2008; Yaffe et al. 1998). Neurosteroids produced by activated microglia are able to shift a pro-inflammatory immune response into an anti-inflammatory phenotype (Giatti et al. 2012). The shift between pro- and anti-inflammatory effects of estrogens seems to be dependent upon both the expression level and the extent of stimulation of estrogen receptors (ER) in the CNS. It is also evident from recent literature that the anti-inflammatory effects are associated with suppression of ER-mediated pro-inflammatory cytokine and chemokine production (Benedusi et al. 2012; Brown et al. 2010).
14.2.2 Progestins
Progestins have been demonstrated to play an important role in neuroprotection as observed in experimental models and clinical trials in patients with stroke and traumatic brain injury (Stein 2011a, b). Progestins can easily pass through the blood-brain barrier and exert their neuroprotective effects inside the brain. A few studies were performed to assess the role of PR in experimental animal models of stroke and traumatic brain injury. Liu et al. found that ischemia to the brain for 6 h resulted in a rapid increase in the progesterone and 5alpha-dihydroprogesterone levels both in wild-type and PR knockout mice, suggesting a possible role of progestins in the salvage of neurons at risk (Liu et al. 2012). Changes in steroid hormone levels affected the expression of membrane PR, specifically PR alpha, in rats and mice. Upon treatment with estradiol or progesterone, significant expression of PR alpha was observed in neurons of olfactory bulb, striatum, cortex, thalamus, hypothalamus, septum, hippocampus, and cerebellum, but not on oligodendrocytes or astrocytes. Traumatic brain injury induced the expression of PR alpha not only on neurons but also on oligodendrocytes, astrocytes, and reactive microglia, suggesting a role of progestins and PR in inflammation in the injured brain (Meffre et al. 2013).
The neuroprotective mechanisms and anti-inflammatory effects of progestins have been reviewed by several authors (Giatti et al. 2012; Luoma et al. 2012; De Nicola et al. 2009; Sayeed and Stein 2009; Singh and Su 2012; Stein et al. 2008). Progestins may prevent brain damage by controlling edema formation (vasogenic or cytogenic) via modulation of the expression of the aquaporin-4 water transporter, moderating Ca2+ flux caused by excitotoxicity, and reconstitution of the blood-brain barrier. Progestins also have antioxidant properties that can prevent cellular insults by oxidative stress induced by free radical formation. Progestins can inhibit the activation of microglia, which prevents NO and TNF-alpha production and the release of other inflammatory cytokines, such as IL-1 beta, TNF-alpha, and IL-6, compliment factor C3 and C5, and macrophage-inducing factor-1. In addition, progestins have anti-apoptotic properties. Besides the aforementioned roles in neuroprotection, progestins also play a role in neuronal remodelling by upregulating several neurotrophic factors, such as brain-derived neurotrophic factor (BDNF), Na/K ATPase, microtubule-associated protein 2 (MAP-2), choline acetyltransferase (ChAT), and glial-derived neurotrophic factor (GDNF).
To investigate progestin as neuroprotectant in patients, a clinical trial (phase IIa) was conducted in 100 male and female patients with blunt head trauma with moderate-to-severe damage. Treatment with progesterone showed significant reduction in mortality compared to vehicle group (Wright et al. 2007). Two large-scale studies on progesterone treatment are now in process. A phase III clinical trial investigates the effects in moderate-to-severe traumatic brain injury in 1,200 patients. Another phase III trial studies the effect on brain injury in pediatric patients (Stein 2011b). Administration of progestins in combination with estrogens was found to be an effective treatment for postmenopausal symptoms. The combination of progestins and estrogens showed efficacy in the treatment of multiple sclerosis in both animal models and in patients. The beneficial effect was mediated by modulation of peripheral and brain-intrinsic immune responses and regulation of local growth factor supply, oligodendrocytes, and astrocytes (Kipp et al. 2012).
14.2.3 Androgens
The most common central effect of androgens is the induction of aggression. Excessive levels of testosterone are known to induce aggression in both sexes. Exposure to high testosterone levels at young age is associated with a reduction in feminine characteristics in women. Moreover, testosterone is converted to estradiol in the CNS, which plays a pivotal role in the feedback regulation in the hypothalamus. The hypothalamus plays a positive role in hormone release by endocrine system through the pituitary. In analogy to menopause in women, aging men may experience andropause (hypogonadism), which is a condition characterized by very low circulating androgen concentrations. This reduction in androgen levels may be accompanied by decreased cell survival in the hippocampus (Spritzer and Galea 2007) and a decrease in cognitive functions and increased risk to develop depression and Alzheimer’s disease (Irie et al. 2006; Seidman 2007). Testosterone treatment was found to reduce many mood- and cognition-related symptoms in hypogonadal men (Zitzmann 2006) and to enhance hippocampal neurogenesis through increased cell survival in rodents (Spritzer and Galea 2007). Androgen treatment combined with estrogens in postmenopausal women provides more improvement in psychologic and sexual symptoms than does estrogen alone (Sarrel 1999).
14.2.4 Corticosteroids
Corticosteroids, such as glucocorticoids and mineralocorticoids, are produced by the adrenal glands, liver, and during pregnancy by placenta and maternal glands. In stressful conditions, the glucocorticoid, cortisol, is rapidly synthesized and secreted in response to adrenocorticotropic hormone released from the pituitary and corticotrophin-releasing hormone secreted by the hypothalamus. Cortisol stimulates the production of energy-rich compounds such as glucose, free fatty acids, and amino acids. The mineralocorticoid aldosterone is produced in response to angiotensin II and promotes sodium reabsorption and fluid retention. Besides their role in glucose and mineral metabolism, corticosteroids are also implicated in the regulation of sleep, ingestive behavior, behavioral adaptation, learning, and memory. In addition, corticosteroid receptors play a significant role in brain damage, aging (De Kloet et al. 1986), mood, mental performance, and the pathogenesis of neuropsychiatric disorders, such as depression and Alzheimer’s disease (McEwen and Sapolsky 1995; Sapolsky 2004). Both cortisol and aldosterone release are controlled by the hypothalamic-pituitary-adrenal (HPA) axis. The HPA axis is a neuroendocrine system that has complex interactions with brain serotonergic, noradrenergic, and dopaminergic systems. An important role of the HPA axis is the regulation of the body’s response to stress. Overactivity of the HPA axis or enlargement of the pituitary or adrenal gland causes an increase in cortisol levels, which in turn leads to hypercortisolemia. The overactivity of the HPA axis in stressful conditions may lead to dysregulation of the serotonergic system and is one of the most important predictors of suicide attempts in depressed patients (Pompili et al. 2010). Depression, on the other hand, is one of the major causes of hypercortisolemia, characterized by increased cortisol levels. Hypercortisolemia may lead to neurotoxicity and reduced neurogenesis in the hippocampus in depressed patients (Sapolsky 1996).
14.3 Steroid Hormone Receptors in Brain Disorders
Steroid hormones exert their biological effects through specific steroid hormone receptors (SHR) that are expressed by the target cells. To date, two estrogen receptor subtypes (ER alpha and ER beta) with several isoforms (Koike et al. 1987; Kuiper et al. 1996; Ma 2000; Menuet et al. 2002), two progesterone receptors (PR) subtypes (PRA and PRB) with several splice variants (Brinton et al. 2008; Misrahi et al. 1987), and two types of androgen receptor (AR) subtypes have been identified (ARA and ARB) (Chang et al. 1988; Wilson and McPhaul 1994). The corticosteroid receptors may be divided into two classes: mineralocorticoid receptors (MR) (Arriza et al. 1987) and glucocorticoid receptors (GR) (Hollenberg et al. 1985). All SHR share similar functional domains but differ in the length of the amino acid chain (Fig. 14.2).
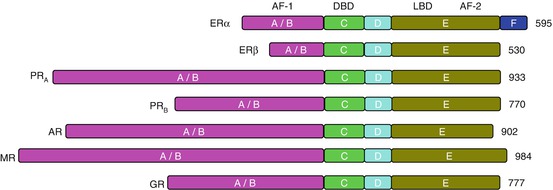
Fig. 14.2
Functional domains of steroid hormone receptors (SHR). All the SHRs consists of common structural domains, in particular an N-terminal transcription activation-1 domain (A/B), which regulates the ligand-independent transcription; a DNA binding domain (C) which recognizes and binds with the steroid hormone response elements on DNA; a flexible hinge domain (D) which carries information for the posttranscriptional modification; and a multifunctional ligand binding domain (E) which recognizes and binds the ligand and acts as a ligand-dependent transcription activation function. Numbers represent the number of amino acids in each receptor. ER alpha contains an extra region (F). ER estrogen receptor, PR progesterone receptor, AR androgen receptor, MR mineralocorticoid receptor, GR glucocorticoid receptor, AF activation function, DBD DNA-binding domain, LBD ligand binding domain
In steroid-responsive cells, SHR are mainly present in the cytoplasm and the nucleus, although SHR are also found on the cell membrane (Jensen et al. 1968; Walsh et al. 1990). When SHR are activated by the corresponding steroid hormone, they dimerize and move into the nucleus of the cell, where they bind to hormone-responsive element in the promoter region of specific target genes. SHR may act as transcriptional activators or transcriptional repressors, resulting in the induction or suppression of the expression of hormone-responsive genes. These responsive genes may evoke a wide variety of physiological responses. Some rapid actions of estrogens and progestins are directly mediated by cell surface receptors in a non-genomic manner (Fig. 14.3). Although the involvement of steroid hormones in various brain disorders has been demonstrated, the mechanisms with which they exert their effects are still largely unknown.
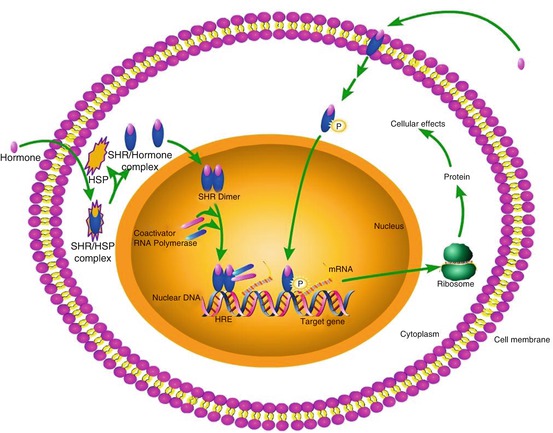
Fig. 14.3
Mode of action of steroid hormone receptors. All the steroid hormone receptors mediate their action by a common genomic pathway. In the cytoplasm, they exist as complexes with heat shock proteins in an inactive form. When a steroid hormone binds, the receptor complex releases the heat shock proteins and becomes activated. The activated receptor forms a homodimer or heterodimer, which subsequently enters into the nucleus. The dimer binds to the specific hormone response element in DNA and initiates or inhibits the process of gene transcription
Western blotting and immunohistochemical studies in experimental animals and postmortem human brains have shown that receptors for different steroid hormones are expressed in different regions of the brain. ER, PR, and AR are expressed in those brain areas that are associated with emotion, cognition, and behavior, such as the hypothalamus, amygdala, cerebral cortex, hippocampus, and brainstem. This expression pattern is in agreement with the association of sex hormones with psychiatric disorders. The neuroprotective effects of estrogens are mainly due to ER alpha-mediated signaling (Ooishi et al. 2012), whereas the effect on mood and cognitive functions in depression and schizophrenia is mainly mediated by ER beta (Foster 2012). A study on patients with depression or schizophrenia, who committed suicide, showed downregulation of ER beta expression in the limbic system (Ostlund et al. 2003). To our knowledge, no data exist on altered expression of PR and AR in the human brain in mood and behavioral disorders.
Not only SHR expression, but also polymorphisms in the SHR genes were found to be implicated in psychiatric diseases. SHR polymorphisms have been associated with an increased risk for schizophrenia, depression, anxiety traits, and cognitive impairment. Association of polymorphisms in estrogen and androgen receptors associated with psychiatric disorders has been reviewed recently by Westbert and Eriksson (2008).
Corticosteroids affect behavioral changes by engaging with either MR or GR. MR are mainly expressed in the limbic system, hypothalamus, and circumventricular organs and to lesser extent in other parts of the brain. The expression of GR, on the other hand, is mainly observed in the subfields of the cerebral cortex, olfactory cortex, hippocampus, amygdala, dorsal thalamus, hypothalamus, cerebellar cortex, trapezoid body, locus coeruleus, and dorsal raphe nucleus in rats (Morimoto et al. 1996). Both the corticosteroid receptors are co-expressed in the hippocampus, amygdala, inferior frontal gyrus, cingulate gyrus, and nucleus accumbens in human brain, whereas predominantly MR were found in the hippocampus (Klok et al. 2011). A reduction in both MR and GR expression and an increase in corticosteroid levels were observed in the brains of patients with bipolar disorder or schizophrenia who committed suicide and in subjects exposed to stressful conditions (Xing et al. 2004). Furthermore, decreased MR expression in the hippocampus of suicide victims was observed in another study (López et al. 1998). Decreased MR expression was also demonstrated in depressed subjects after treatment with the MR antagonist, spironolactone. Spironolactone treatment led to an increase in cortisol levels in both controls and depressed patients. However, in depressed patients, cortisol levels were significantly higher than in controls (Young et al. 2003).
Increased production of the cortisol also causes desensitization of the GR, resulting in a downregulation of these receptors. Downregulation of GR in turn leads to an increase in the levels of cortisol (Hansen-Grant et al. 1998). Prolonged stress has been reported to downregulate the expression of GR in the prefrontal cortex in rats (Chiba et al. 2012). Likewise, stress leads to a reduction in the GR mRNA in the basolateral/lateral nuclei in patients with schizophrenia or bipolar disorder (Perlman et al. 2004, 2007).
14.4 Imaging of Steroid Hormone Receptors
Hitherto, almost all information about the role of SHR in healthy and diseased brain is obtained from experimental animals and postmortem human studies, because brain biopsy in patients is generally not feasible or highly undesirable. To better understand the role of steroid hormones in neurodegenerative and psychiatric diseases, it is therefore of invaluable importance to have a noninvasive tool to measure the expression of SHR in the brain. PET and SPECT are noninvasive nuclear imaging techniques that allow measurement of receptor expression and receptor occupancy in the living brain. Until now, several PET and SPECT tracers have been developed to image the SHR. Most of these radiopharmaceuticals, however, were developed for applications in oncology, in particular for imaging of receptor expression and occupancy in steroid hormone-sensitive tumors like breast and prostate cancer (Hospers et al. 2008; De Vries et al. 2007). So far, only a few studies on imaging of SHR in the brain have been reported. However, it is expected that most radiopharmaceuticals that have been developed for tumor imaging may also be applied in brain imaging. Although many tracers have been described for each SHR, most of these tracers did not provide satisfactory results and consequently did not enter clinical studies. In the following paragraphs, we will not provide a complete overview of all tracers for SHR that have been reported, but only discuss the most promising candidate tracers for PET and SPECT imaging of SHR in the brain.
14.4.1 Radiopharmaceuticals for Estrogen Receptor Imaging
ER is the most widely studied SHR. Although literature exists on Western blotting and in situ hybridization studies to determine the expression of the ER in the rodent brain, hardly any data exists on ER expression in the human brain. For this purpose, imaging methods to measure ER expression could certainly be of added value. Several tracers have been developed for PET and SPECT imaging of ER, in particular for imaging breast cancer.
14.4.1.1 PET
16Alpha-[18F]fluoro-17beta-estradiol ([18F]FES) was introduced for imaging of ER in the 1980 and is now used in both clinical trials and in patient care for diagnosis and monitoring of anticancer treatment efficacy in ER-positive breast cancer. In rats and mice, highest [18F]FES uptake was observed in tissues with high ER expression, such as the uterus and ovaries (Aliaga et al. 2004; Kiesewetter et al. 1984; Sasaki et al. 2000; Seimbille et al. 2002; Yoo et al. 2005). [18F]FES uptake in ER-rich organs could be blocked with unlabeled estradiol in a dose-dependent manner, indicating that the tracer uptake is ER-mediated (Katzenellenbogen et al. 1993). A recent study demonstrated that treatment with fulvestrant, an irreversible ER antagonist, also reduced the uptake of [18F]FES in ER-positive breast tumors in mice (Fowler et al. 2012). PET imaging studies in rodents showed a good correlation of [18F]FES tumor uptake with ER density, as determined in vitro (Aliaga et al. 2004). In breast cancer patients, [18F]FES PET has been applied successfully to determine the ER status of tumor lesions. ER expression was clearly visualized in primary breast tumors and in metastases. The accumulation of [18F]FES in these tumors correlated well with ER density, as determined by immunohistochemistry (Dehdashti et al. 1995; Mintun et al. 1988). The radiation burden associated with [18F]FES PET is within the normal range of other clinical nuclear medicine procedures: a typical dose of 200 MBq (6 mCi) causes a radiation burden to the patient of 4.4 mSv (Mankoff et al. 2001; Mortimer et al. 1996; Peterson et al. 2008). [18F]FES preferentially binds to the alpha subtype of the ER, as its affinity is 6.3-fold higher for ER alpha than for ER beta (Yoo et al. 2005). ER alpha is overexpressed in many breast tumors, where it is associated with tumor growth and development. The ER beta isoform was found to be co-expressed with ER alpha in breast tumors (Dotzlaw et al. 1997; Järvinen et al. 2000), but the function of ER beta in breast cancer is not well understood yet. The discovery of ER beta in breast cancer initiated the search for ER beta-selective PET tracers. Several attempts have been made to develop ER beta-selective tracers. Recently, 8beta-(2-[18F]fluoroethyl)estradiol (8beta-[18F]FEE), a derivative of the potent ER beta-selective steroid 8beta-vinylestradiol, was evaluated as a selective PET tracer for ER beta. In addition, the nonsteroidal ER beta ligand ERB-041 was labeled with 76Br, yielding [76Br]BrERB-041 as a potential ER beta-selective PET tracer. Although both 8beta-[18F]FEE and [76Br]BrERB-041 showed high binding affinities in a radiometric assay, ex vivo biodistribution studies in rats and mice could not demonstrate ER beta-mediated uptake (Lee et al. 2012). So far, no suitable PET tracer of imaging of ER beta is available.
Besides [18F]FES, several radiolabeled estradiol derivatives and ER-targeting anticancer drugs have been evaluated as candidate PET tracers for imaging ER density and occupancy (Fig. 14.4). The potent estrogen 17alpha-ethynyl-11beta-methoxy-estradiol ([18F]betaFMOX) showed promising results in rodents with a fourfold higher uptake in immature rat uterus than [18F]FES. [18F]betaFMOX was considered to be a highly sensitive tracer for imaging of the ER, as [18F]betaFMOX also exhibited specific binding in organs with low ER density, such as the kidney, muscle, and thymus (VanBrocklin et al. 1993a, b). Despite the promising results in rodents, [18F]betaFMOX PET studies failed to detect ER-positive lesions in human breast cancer patients. This lack of sensitivity in humans may be due to the fast metabolism of the tracer, because it exhibited low binding to sex hormone-binding protein (SHBG), which protects steroids from metabolic degradation (Jonson et al. 1999). Other ethynyl analogues of FES also showed better target-to-background ratios but were also associated with faster degradation in vivo, because of low SHBG binding (VanBrocklin et al. 1992).
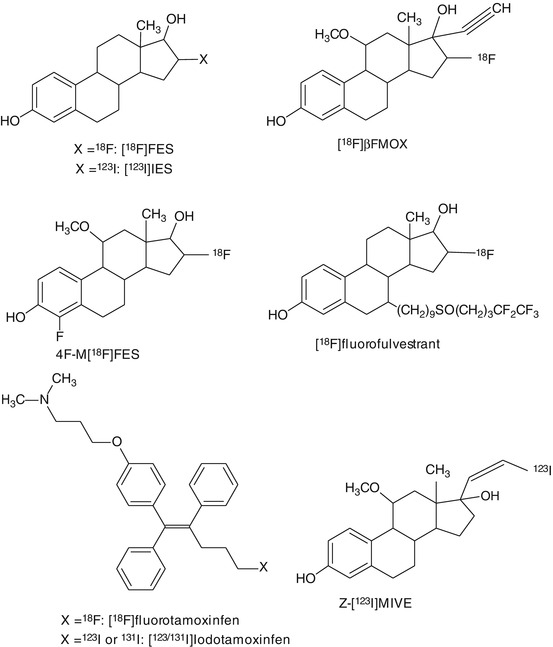
Fig. 14.4
Chemical structures of tracers that have been evaluated for PET or SPECT imaging of estrogen receptors
In order to increase the binding affinity and in vivo stability of the tracer, several analogues of [18F]FES were developed. One of these candidates, 11beta-methoxy-4,16alpha-[18F]difluoroestradiol (4F-M[18F]FES), showed high uptake and high target-to-background ratios in ER-rich organs like the uterus (VanBrocklin et al. 1994). 4F-M[18F]FES has a low affinity towards the SHBG, but studies in humans showed a significant, potentially ER-mediated uterus uptake in both pre- and postmenopausal women (Beauregard et al. 2009). However, no other human studies with 4F-M[18F]FES have been published since.
In addition to estradiol derivatives, radiolabeled derivatives of anticancer drugs, such as tamoxifen and fulvestrant, were evaluated for imaging of ER. Tamoxifen is a partial agonist of the ER and used as a drug in the treatment of ER-positive breast cancer. 18F-labeled tamoxifen, [l8F]fluoromethyl-N,N-dimethyltamoxifen ([I8F]FTX), exhibited specific uptake in the uterus and mammary tumors, which could be blocked with an excess of estradiol (Yang et al. 1994). In a clinical study in 10 ER-positive breast cancer patients, [I8F]FTX uptake appeared to correlate with tamoxifen treatment outcome (Inoue et al. 1996). So far, no additional studies have been performed to prove the clinical utility of [I8F]FTX. Labeling of fulvestrant, a full antagonist of the ER, with 18F reduced its binding affinity. Consequently, 18F-labeled fulvestrant is not suitable for PET imaging of ER (Seimbille et al. 2004).
14.4.1.2 SPECT
In addition to PET tracers, some SPECT tracers were developed for imaging of ER (Fig. 14.4). Amongst the promising SPECT tracers for ER was 16alpha-[125I]iodo-11beta-methoxy-17beta-estradiol ([125I]MIE) (Zielinski et al. 1986). In rodents, [125I]MIE was found to accumulate in ER-rich areas, such as the uterus. [125I]MIE showed higher uterus-to-blood ratios than 16alpha-[125I]iodoestradiol. 16alpha-[123I]iodo-17beta-estradiol ([123I]IES) and (20Z)-11beta-methoxy-17alpha-[123I]iodovinylestradiol (Z-[123I]MIVE) have not only been applied in animals but also in clinical studies. [123I]IES showed ER-mediated uptake in the rabbit reproductive system and in ER-positive tumors in breast cancer patients, but no specificity or sensitivity data were reported (Kenady et al. 1993; Pavlik et al. 1990; Scheidhauer et al. 1991; Schober et al. 1990). Both stereoisomers of [123I]MIVE were evaluated for ER imaging. (Z)-[123I]MIVE showed highest ER-mediated specific uptake in rodents and in clinical studies in breast cancer patients. The tracer uptake could be blocked by saturation of the ER with tamoxifen (Bennink et al. 2001, 2004; Rijks et al. 1997a, b). Although both [123I]IES and (Z)-[123I]MIVE showed ER-mediated specific uptake, these tracers also displayed fast metabolism and low SHBG binding (Nachar et al. 1999; Rijks et al. 1998b). For both tracers, no further studies are available to support their utility for brain imaging.
Attempts have been made to generate a SPECT tracer for ER by labeling derivatives of the anticancer drug tamoxifen. In rodents, the uptake of [123I]iodotamoxifen ([123I]TAM) was found to be high in ER-rich organs. In clinical studies, [123I]TAM showed ER-specific uptake in breast tumors (Hanson and Seitz 1982; Van de Wiele et al. 2001). Recently tamoxifen was also labeled with 131I ([131I]TAM). Biodistribution studies in rodents proved that the uptake of this tracer was ER-mediated in the uterus and breast tissue (Muftuler et al. 2008). No further studies on iodine-labeled tamoxifen have been published lately.
Estradiol and anticancer drug derivatives have also been labeled with 99mTc. A few of them have even been successfully evaluated in rodents (Arterburn et al. 2003; Nayak et al. 2008; Ramesh et al. 2006; Takahashi et al. 2007a, b; Yurt et al. 2009; Zhu et al. 2010). However, the usefulness of these tracers for brain imaging would be questionable, because of the high hydrophilicity of the 99mTc conjugates, which likely precludes efficient brain penetration.
Consequently, at this moment [18F]FES remains the only validated tracer for molecular imaging of ER expression that is currently used in clinical studies. Even though its characteristics are not ideal, [18F]FES still is an adequate tracer for PET imaging of ER in humans (Van Kruchten et al. 2012). Efforts to develop better alternatives for [18F]FES are still made but yielded disappointing results so far.
14.4.2 Radiopharmaceuticals for Progesterone Receptor Imaging
14.4.2.1 PET
A few substrates of PR have been labeled with 18F, 76Br, 123I, and 125I for imaging of PR with PET and SPECT (Fig. 14.5). The 18F-labeled candidate PET tracers for PR include 21-[18F]fluoro-16alpha-ethyl-19-norprogesterone ([18F]FENP) (Dehdashti et al. 1991; Jonson and Welch 1998; Pomper et al. 1988; Verhagen et al. 1991a), 21-[18F]fluoro-16alpha-methyl-19-norprogesterone ([18F]FMNP) (Verhagen et al. 1991b), and 21-[18 F]fluoro-16alpha,7alpha-[(R)-(1′alpha-furylethylidene)dioxy]-19-norpregn-4-ene-3,20-dione (Buckman et al. 1995). [18F]FMNP exhibited PR-mediated uptake in the uterus and tumors in rats (Verhagen et al. 1991b), but no further studies in humans were reported. [18F]FENP showed highly selective PR-mediated uptake in the uterus of estrogen-primed rats (Pomper et al. 1988) and in PR-positive carcinoma in mice (Verhagen et al. 1991a). In eight breast cancer patients, however, [18F]FENP could only detect 50 % of PR-positive lesions, and tracer uptake did not correlate with PR expression (Dehdashti et al. 1991). The major reason for failure of [18F]FENP in clinical studies was its extensive metabolism in humans (Verhagen et al. 1994). To overcome this problem, several ketals of 16alpha,17alpha-dihydroxyprogestrerone were labeled with positron-emitting isotopes (Buckman et al. 1995; Vijaykumar et al. 2002; Zhou et al. 2006). One of these ketals is 21-[18F]fluoro-16alpha,17alpha-[(R)-(1′-alpha-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione ([18F]FFNP). In a study performed in mice with mammary tumors, treatment with estradiol, letrozole, or fulvestrant showed treatment-induced changes in the uptake of [18F]FFNP, suggesting that early evaluation of response to the anticancer treatment could be feasible (Fowler et al. 2012). Recently, a study in breast cancer patients showed that [18F]FFNP PET is a safe, noninvasive method to evaluate the tumor’s PR status in vivo (Dehdashti et al. 2012). Brain uptake of [18F]FFNP seems to be acceptable, but further studies are needed to prove its suitability for brain imaging.
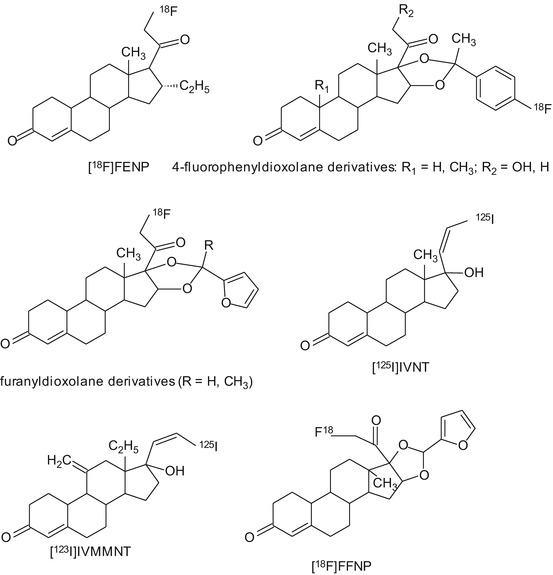
Fig. 14.5
Chemical structures of tracers that have been evaluated for PET or SPECT imaging of progesterone receptors
Another series of candidate PET tracers for PR imaging were the derivatives of the nonsteroidal PR agonist tanaproget. Several derivatives of tanaproget were evaluated (Zhou et al. 2010), and 4-[18F]fluoropropyl-tanaproget ([18F]FPTP) demonstrated highest uptake in the target tissues like uterus and ovaries. The biodistribution pattern of [18F]FPTP in rats was comparable with other PR tracers, such as FENP and FFNP (Lee et al. 2010).
14.4.2.2 SPECT
A few iodinated tracers for PR imaging have been developed, but none of these candidate tracers showed promising results. (20Z)-17alpha-[125I]iodovinyl-19-nortestosterone ([125]IVNT) had interesting binding properties but lacked selectivity in vivo (Ali et al. 1994). Later, both isomers of 17alpha-iodovinyl-18-methyl-11-methylene-19-nortestosterone ([123I]IVMMNT) were investigated in rats and rabbits. (Z)-[123I]IVMMNT displayed highest in vivo binding in target organs, such as in uterus and ovaries. The uptake of (Z)-[123I]IVMMNT was found to be PR-mediated (Rijks et al. 1998a). So far, no human data exist to prove the feasibility of PR imaging with these tracers.
At the moment, [18F]FFNP seems to be the most promising candidate tracer for imaging PR, but more studies are required to validate the utility of this tracer in humans.
14.4.3 Radiopharmaceuticals for Androgen Receptor Imaging
14.4.3.1 PET
The existing PET tracers for imaging AR expression are depicted in Fig. 14.6. 20-[18F]fluoromibolerone (20-[18F]Fmib) was the first radiolabeled PET tracer for AR that showed promising uptake in the prostate of diethylstilbestrol (DES)-primed rats (Liu et al. 1991). Several other fluorinated compounds have been developed, and some of them showed promising results, such as 16beta-[18F]fluorodihydrotestosterone ([18F]FDHT), 16beta-[18F]fluorotestosterone (16beta-[18F]FT), 16beta-[18F]fluoro-7alpha-methyl-19-nortestosterone (16beta-[18F]FMNT), 16alpha-[18F]fluoro-7alpha-methyl-19-nortestosterone (16alpha-[18 F]FMNT), and 20-[18F]fluorometribolone (20-[18F]R1881) (Liu et al. 1992). Most of these tracers showed quick clearance from the body and rapid metabolism. Best results in baboons and rats were obtained for [18F]FDHT. Uptake of [18F]FDHT in the prostate was specific and AR-mediated. Metabolism of [18F]FDHT was slower than metabolism of the other candidate tracers (Bonasera et al. 1996; Choe et al. 1995). [18F]FDHT was able to detect AR-positive tumor lesions in prostate cancer patients, and tracer uptake was proven to be AR-mediated in humans as well (Dehdashti et al. 2005; Larson et al. 2004). Another interesting candidate PET tracer is 7alpha-[18F]fluoro-17-methyl-5-dihydrotestosterone ([18F]FMDHT), although the first results reported for this tracer were not very promising. The uptake of [18F]FMDHT in the prostate was low; results were not reproducible and not comparable with other labeled steroids like [18F]FDHT. In these first experiments, [18F]FMDHT was investigated in rats treated with DES to suppress endogenous testosterone production (Garg et al. 2001). The same authors reevaluated the suitability of [18F]FMDHT for AR imaging in chemically castrated rats. In this animal model, uptake in the prostate was proven to be AR-mediated. Prostate uptake was comparable to other 18F-labeled steroids and twofold higher than [18F]FMDHT uptake in DES-treated rats (Garg et al. 2008).
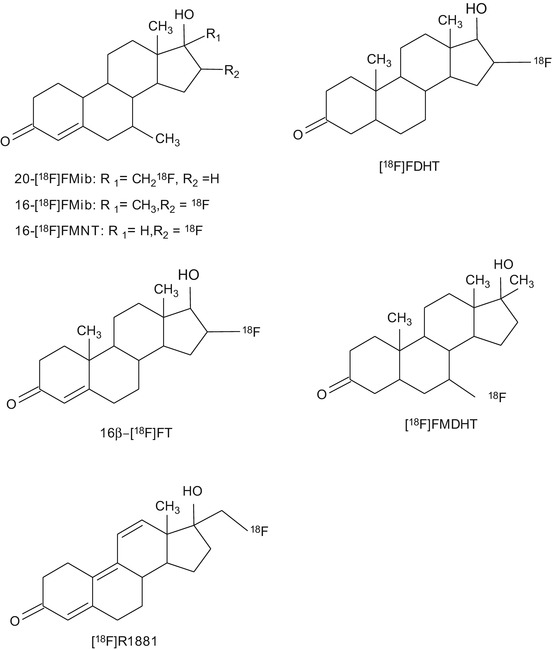
Fig. 14.6
Chemical structures of tracers that have been evaluated for PET or SPECT imaging of androgen receptors
Currently, [18F]FDHT is the only PET tracer that has proceeded into the clinical evaluation phase (Beattie et al. 2010; Dehdashti et al. 2005; Larson et al. 2004; Zanzonico et al. 2004). The radiation burden associated with [18F]FDHT PET is within the normal range of other clinical nuclear medicine procedures: 0.018 mSv/MBq; for the maximum administered dose of 331 MBq, the total radiation burden is 6.0 mSv. The major drawback of [18F]FDHT is its rapid metabolism. This led to the investigation of nonsteroidal derivatives with better in vivo stability, like propanamide derivatives a selective androgen receptor modulator (SARM) with 11C (Gao et al. 2011).
Some nonsteroidal antagonists of AR have been radiolabeled, such as a 11C-labeled diethylamine flutamide derivative (Jacobson et al. 2006), a 18F-labeled hydroxyflutamide derivative (Jacobson et al. 2005), 3-[76Br]bromohydroxyflutamide (Parent et al. 2006), [18F]bicalutamide, 4-[76Br]bromobicalutamide, and [76Br]bromo-thiobicalutamide (Parent et al. 2007). However, none of these compounds showed promising results that warranted further evaluation.
14.4.3.2 SPECT
The radioiodinated steroid 2alpha-[125I]dihydrotestosterone was the first SPECT tracer showing high uptake in AR-rich organs like the prostate, epididymis, and testis in rats. Pretreatment with dihydrotestosterone reduced the uptake in these organs, suggesting that tracer uptake is specific and AR-mediated (Tarle et al. 1981). 7alpha-[125I]iodo-5alpha-dihydrotestosterone (7alpha-[125I]IDHT) is another analogue of testosterone that was labeled with 125I. 7alpha-[125I]IDHT showed AR-mediated uptake in the prostate of rats. In vitro autoradiography of 7alpha-[125I]IDHT produced excellent autoradiograms with low nonspecific binding in the prostate of rats (Labaree et al. 1997), but no further studies were published to demonstrate the use of this tracer in vivo.
Some steroid and flutamide derivatives have been labeled with 99mTc and tested as SPECT tracers for AR. However, in vivo evaluation in rats did not show any AR-mediated specific binding for any of these compounds (Dallagi et al. 2010; Dhyani et al. 2011).
So far, [18F]FDHT is the only AR tracer that has proceeded into the clinical evaluation phase. Clinical studies confirmed that this tracer is suitable for AR imaging in cancer patients. However, it still remains to be evaluated whether [18F]FDHT PET is also able to monitor AR expression in the human brain. Preclinical data of [18F]FMDHT suggest that it might have favorable characteristics for AR imaging as well, but further evaluation is still required to establish the merit of this tracer.
14.4.4 Radiopharmaceuticals for Corticoid Receptor Imaging
14.4.4.1 Radiopharmaceuticals for Glucocorticoid Receptor Imaging
So far, only a few GR ligands have been synthesized and evaluated for the potential use as imaging tracer (Fig. 14.7). Most of these candidate tracers showed disappointing results in rodents and baboons. The first labeled compound that was introduced as ligand for the imaging of GR was 21-[18F]fluoroprednisone. 21-[18F]fluoroprednisone was rapidly metabolized, leading to low uptake in rat brain (Feliu and Rottenberg 1987).
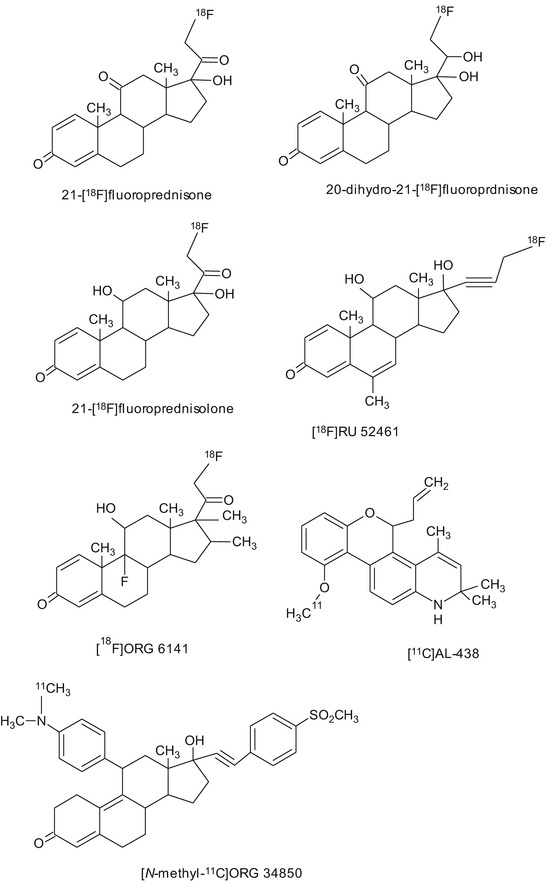
Fig. 14.7
Chemical structures of tracers that have been evaluated for PET or SPECT imaging of glucocorticoid receptors
The biodistribution of [18F]RU 52461, an analogue of the selective GR agonist RU28362, showed high GR-mediated uptake in the adrenals and pituitary in rats. PET imaging studies in baboons, however, showed low uptake of [18F]RU 52461 in the brain (Dasilva et al. 1992). In another study in rats, [18F]RU 52461 uptake in the hippocampus could only be partially blocked with an excess of the unlabeled ligand, whereas complete blocking of tracer uptake in peripheral organs was observed (Pomper et al. 1992).
The potent GR ligand [18F]ORG 6141 was evaluated in adrenalectomized and sham-operated rats. Ex vivo biodistribution 3 h postinjection revealed higher uptake of tracer in the hippocampus and brain stem of adrenalectomized animals as compared to sham-operated controls. However, GR-mediated specific retention of activity in these brain areas could not be demonstrated (Visser et al. 1995).
Wuest et al. synthesized a series of novel 4-fluorophenylpyrazolo steroids and tested their binding affinities to GR. Some of these compounds showed binding affinities up to 56 % relative to dexamethasone (100 %) (Wüst et al. 2003). One of these compounds, 2′-(4-fluorophenyl)-21-[18F]fluoro-20-oxo-11beta,17alpha-dihydroxy-pregn-4-eno[3,2-c]pyrazole, was evaluated in rats using autoradiography and small animal PET imaging. Brain uptake of this tracer was found to be constant between 5 and 60 min after tracer injection. However, brain uptake was not specifically GR-mediated (Wüst et al. 2005), but only due to nonspecific binding.
The high-affinity GR antagonist Org 34850 was labeled with 11C. [N-methyl-11C]Org 34850 was found to rapidly metabolize in rats. Ex vivo biodistribution and small animal PET studies demonstrated that [N-methyl-11C]Org 34850 was not able to penetrate the blood-brain barrier (Wuest et al. 2009).
The nonsteroidal selective GR modulator AL-438 was labeled with 11C. The biodistribution of [11C]AL-438 showed high uptake in the pituitary and the brain, but treatment with a high dose of the GR antagonist corticosterone did not result in any blocking of tracer uptake, suggesting that the uptake was not GR-mediated (Wuest et al. 2007).
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Tracers for Beta-Amyloid and Other Proteinopathies
 PET Imaging of ABC Transporters in the BBB
PET Imaging of ABC Transporters in the BBB
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


