CNS disease
ABC transporters examined in the context of this disease
References
Ischemic stroke
ABCB1, ABCC1
Epilepsy
ABCB1, various ABCCs, ABCG2
Brain cancer
ABCB1, ABCC1, ABCC4, ABCC5, ABCG2
Parkinson’s disease, progressive supranuclear palsy, multiple system atrophy
ABCB1
Alzheimer’s disease
ABCA1, ABCA2, ABCB1, ABCC4, ABCG2
Adrenoleukodystrophy
ABCD1
Multiple sclerosis
ABCB1, ABCG2
Brain metabolic disorders
ABCA1, ABCA2, ABCC8 (SUR1), ABCC9 (SUR2), ABCG1, ABCG4
As polymorphisms of these genes are critical for prediction of neurological diseases, their pharmacogenetic data is also of great value. The ABCB1 gene contains more than 50 single nucleotide polymorphisms (SNP), some of them linked to changes in the expression and/or function of this ABC transporter (Sharom 2007). However, the difficulty in replicating results associating polymorphisms with the risk of developing diseases complicates this area of research. For example, many studies have investigated the relationship between ABCB1 polymorphisms and the risk of developing Parkinson’s disease (PD), but to date only few studies have established such a direct association (Droździk et al. 2003; Lee et al. 2004; Westerlund et al. 2009). Therefore, ABCB1 variants are now thought to act rather indirectly as a susceptibility factor or modulator of the risk of PD in conjunction with the exposure to known risk factors such as pesticides (Brown et al. 2006). Noteworthy, three studies of genetic polymorphisms related to P-gp have underscored the increased risk of developing PD for carriers of variant C3435T or G2677[A,T] ABCB1 alleles who have also been exposed to pesticides (Droździk et al. 2003; Dutheil et al. 2010; Zschiedrich et al. 2009).
In relation to Alzheimer’s disease (AD), ABCB1 and ABCG2 have recently been linked to the transport of Aβ peptides at the BBB. However, Vogelgesang et al. found no significant association between ABCB1 polymorphisms in humans and the risk of AD, but they demonstrated an inverse correlation between Aβ deposition rates and the cerebrovascular ABCB1 expression (Vogelgesang et al. 2002, 2004). Another recent study showed that besides the age-related decrease of P-gp expression, the Aβ1-42 peptide itself is able to downregulate the expression of P-gp (Brenn et al. 2011). In contrast, the amount of ABCG2 in the brains of AD and CAA (cerebral amyloid angiopathy) patients was higher than in age-matched controls. Furthermore, the brains of Abcg2-null mice significantly accumulated more Aβ peptides compared to wild-type mice (Xiong et al. 2009), confirming that this transporter plays a protecting role against Aβ overload in the brain. In addition, the implication of both ABCB1 and ABCG2 in preventing Aβ from entering the brain was already confirmed by in vitro studies (Candela et al. 2010; Tai et al. 2009). This coordinated action between these two transporters is now the object of study of several research groups (Agarwal et al. 2011, 2012; Agarwal and Elmquist 2012; Kodaira et al. 2010), which certainly will provide a better understanding of their function at the BBB.
Since many in vitro results about ABC transporters may not be representative for the in vivo situation, functional imaging of these transporters at the BBB with an in vivo PET assay will increase our knowledge on the role of these proteins in physiological and pathophysiological conditions.
It has been established that considerable species differences exist concerning the expression of several ABC transporters in the BBB (Hermann and Bassetti 2007; Soontornmalai et al. 2006). A recent proteomic analysis conducted by Uchida and colleagues in both human and mouse brain microvessels lends support to this conclusion, and several examples illustrate this fact. Expression of BCRP protein was almost twice as high (1.85-fold) in humans as in mice. In contrast, MDR1 and MRP4 protein expressions were smaller in humans than in mice, 2.33- and 8.15-fold, respectively (Uchida et al. 2011). Thus, imaging of ABC transporters and comparison of the data from different animal strains and from animal and human tissues will certainly expand our knowledge in this field. In addition, it will facilitate the translation of pharmacological findings from one animal strain to another, as well as from animals to humans.
21.2 PET
Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) together with magnetic resonance imaging (MRI) and computed tomography (CT) are imaging techniques that allow noninvasive tracking of pathophysiological processes in various neuropsychiatric disorders. Molecular imaging has also been used in various aspects of drug development (Willmann et al. 2008). For an efficient visualization, characterization, and measurement of biological processes at the molecular, cellular, whole organ, or body level, specific imaging probes are needed. These probes are also known as tracers, due to the sub-pharmacological amounts administered. They provide an analytical signal which is detected by a particular method resulting in either a two- or three-dimensional image. PET is the most sensitive technique for in vivo imaging and quantification of biological and pathophysiological processes. However, the spatial resolution of PET is relatively low, and the additional use of CT or MRI imaging is required to provide high-resolution structural images that can be fused with the functional PET image. This will give both anatomical and functional information in a single scanning session.
The decay of these radionuclides occurs by the emission of a positively charged particle called positron. When emitted from the nucleus, the positron travels a very short distance (positron range, 3–5 mm) in the surrounding tissue before it annihilates by combining with an electron. Then, the mass of the positron and electron are converted to energy resulting in two 511 keV γ-rays which are emitted simultaneously at approximately 180° to each other. The pair of γ-rays is detected by rings of detectors in the PET scanner. The acquisition of a large number of coincident events provides data which can be reconstructed to an image with information on the spatial distribution of radioactivity as a function of time.
21.3 P-gp Substrate, Inhibitor, and Modulator Terminology Overview
In relation to P-gp binding (usually a reversible, noncovalent binding), a terminology overview is necessary. When a substrate (sometimes called a P-gp activator) binds to P-gp (in the substrate site of the P-gp molecule) or to another ABC transporter (depending on the molecular affinity), it is pumped out of the cell in an ATP-dependent manner. On the other hand, P-gp inhibitors bind to P-gp (or other ABC transporters, depending on the specificity of the molecule) and then block its activity, preventing P-gp-mediated drug transport and allowing lipophilic molecules to get into the cells. A P-gp modulator also increases the tissue penetration of drugs transported by these proteins, but by a different mechanism of action. Modulators usually bind (noncovalently) to specific regulatory site(s) on the transporter molecule, thus causing a conformational alteration of the P-gp tertiary structure which is known as allosteric modulation (in this case a negative allosteric modulation of the transporter occurs). Modulators can bind to other regulatory site(s) of the P-gp structure as well and cause activation instead of inhibition by a process called positive allosteric modulation. It is important to consider that P-gp substrates such as verapamil and cyclosporine A can also bind to site(s) other than the substrate site, resulting in inhibition of P-gp activity (Maki and Dey 2006). For the more recent second- and third-generation inhibitors, the modes of action are yet to be determined. Based on experimental evidence, there is a growing consensus in favor of an allosteric mode of action for some of these compounds. Independent studies (Dey et al. 1997, 1998, 1999) suggested the existence of distinct modulator site(s) within the P-gp molecule that are linked to substrate site(s) either by negative or positive heterotropic cooperativity (heterotropic when the modulator is a molecule other than the substrate, otherwise it should be called homotropic cooperativity). Finally, it should be noted that conformational changes of the P-gp molecule can be induced by both ligand and ATP (Moaddel and Wainer 2007).
21.4 PET Tracers for Imaging of ABC Transporter Function and Expression in the BBB
21.4.1 Background
Positron emission tomography (PET) is a widely used technique that allows measurement of the expression or function of proteins involved in many neurological diseases including cancer. The most demanding challenge to date is to synthesize the best tracer for a trustworthy clinical diagnosis (Kannan et al. 2009). In relation to ABC transporters, and especially to P-gp, there are three criteria for the selection of the optimal tracer: high radiochemical purity, high selectivity for P-gp, and a large-magnitude signal (Kannan et al. 2009). Combinations of several radiolabeled and nonradioactive P-gp inhibitors are currently used in order to obtain a more specific imaging analysis, allowing the tracer to enter the brain tissue (see text below).
It is important to note that, beyond the scope of diagnosis, P-gp inhibitors have also been widely studied in order to improve treatment outcome in conditions of chemoresistance. Many clinical trials have attempted to overcome the MDR phenomenon by pharmacological inhibition of P-gp (McHugh et al. 2008; McHugh 2008). However, 30 years of such trials have yielded little or no success (Kannan et al. 2009), mainly because of a poor inhibitor selectivity. Other ABC transporters not involved in MDR are affected by the drug treatment which may lead to severe side effects, and the CYP3A4 enzyme that is responsible for metabolism of chemotherapeutic agents is inhibited (van Zuylen et al. 2000). Hence the search for more potent, efficacious, and selective P-gp inhibitors continues, for both diagnostic and treatment purposes.
In theory, P-gp inhibitors can be used to image P-gp expression, whereas P-gp substrates will image P-gp function (Dörner et al. 2011). However, in practice, these compounds do not show the same behavior in vivo and in vitro, which is a great challenge for neuroimaging studies. An important point to consider is membrane fluidity that is influenced by age and diet composition. In addition, sphingolipids can modulate membrane composition and properties through reciprocal interconversions and specific lipid–protein interactions (Posse de Chaves and Sipione 2010). This can alter the structure and spatial conformation of membrane proteins, conferring them distinct ligand-binding aspects.
Several PET tracers have been synthesized for monitoring the function and expression of P-gp at the BBB. Some of them were initially described as P-gp substrates such as [11C]verapamil (Luurtsema et al. 2003) and [11C]N-desmethyl-loperamide (Lazarova et al. 2008). Others are P-gp inhibitors such as [11C]laniquidar (Luurtsema et al. 2009), [11C]elacridar (Dörner et al. 2009; Kawamura et al. 2011a), [11C]tariquidar (Bauer et al. 2010; Kawamura et al. 2010), and [11C]MC18 (van Waarde et al. 2009).
21.4.2 Substrates as Tracers for Measuring P-gp Function
21.4.2.1 [11C]Verapamil
The L-type calcium channel blocker verapamil [2,8-bis(3,4-dimethoxyphenyl)-6-methyl-2-isopropyl-6-azaoctanitrile] is widely used in the treatment of hypertension (McTavish and Sorkin 1989), angina pectoris, and cardiac arrhythmias. Verapamil metabolizes preferentially to norverapamil via N-demethylation (Kroemer et al. 1993). After oral administration, the (S)-enantiomer of verapamil is metabolized more actively than (R)-verapamil in humans, resulting in a 2.5 times higher concentration of the (R)-enantiomer in plasma (Häussermann et al. 1991). (R)-verapamil has only 5–10 of the calcium channel blocking activity of the (S)-enantiomer. However, the (R)-enantiomer is equipotent to racemic verapamil and (S)-verapamil in its efficacy to inhibit MDR. Verapamil is metabolized by cytochrome P450 enzymes and some of the radioactive metabolites are themselves substrates and inhibitors of P-gp (Pauli-Magnus et al. 2000). In addition, polar radiolabeled metabolites are formed, which may result in a non-P-gp-mediated signal (Luurtsema et al. 2005). Quantification of racemic [11C]verapamil (Fig. 21.1) is difficult because the enantiomers have different pharmacokinetic properties (Laberge et al. 2001). Therefore PET studies need to be performed with an optically pure enantiomer. (R)- and (S)-[11C]verapamil were prepared from (R)- and (S)-desmethylverapamil by methylation with no-carrier added [11C]methyl iodide or [11C]methyl triflate (Luurtsema et al. 2002). However, no difference in transport for P-gp between (R)- and (S)-verapamil was found (Luurtsema et al. 2003). (R)-[11C]verapamil had a low brain uptake at baseline both in humans and other vertebrate species, but coadministration with inhibitor cyclosporin A (CsA) or tariquidar increased distribution volume (DV) and kinetic constant k 1 (Bankstahl et al. 2008b; Muzi et al. 2009). The increase in brain DV was approximately +300 % in rats and +24 ± 15 % in humans after administration of tariquidar (Kuntner et al. 2010; Wagner et al. 2009), which points to a pronounced species difference regarding the tariquidar effect. Three different PET tracers, including [11C]verapamil, have been tested in five different mammals. The brain uptake of all radioligands was clearly higher in humans, monkeys, and minipigs than in rats or guinea pigs (Syvänen et al. 2009).
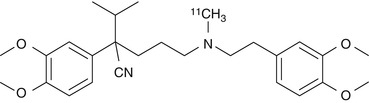
Fig. 21.1
Structure of [11C]verapamil
21.4.2.2 [11C]N-desmethyl-Loperamide
Loperamide is an antidiarrheal agent that acts through agonism of gut μ-opioid receptors (Awouters et al. 1993). This drug is normally without harmful central effects because P-gp expels it from the brain. [11C]Loperamide is avidly metabolized to [N-methyl-11C]N-desmethyl-loperamide ([11C]dLop) via demethylation (Fig. 21.2) (Zoghbi et al. 2008). This metabolite also behaves as a substrate for P-gp and it has been used as a PET tracer instead of [11C]loperamide. Demethylation of [11C]dLop may also occur, but it will lead only to single-carbon radiometabolites such as [11C]methanol. These metabolites will be oxidized and ultimately expired as [11C]carbon dioxide. They should not accumulate in brain tissue and will therefore not cause problems in biomathematical analysis of the acquired PET data. The uptake of [11C]dLop-derived radioactivity at 30 min was about 3.5-fold higher in the forebrain of P-gp knockout mice compared to wild-type mice supporting the results from previous studies which indicate that [11C]dLop is a substrate of P-gp (Lazarova et al. 2008). Similar results were found in monkeys. [11C]dLop was found to be selective for P-gp when tested in human cell lines that overexpress P-gp, MRP1, or BCRP and also in P-gp knockout mice (Kannan et al. 2010). At low concentrations (≤1 nM) dLop acts only as a substrate, but at higher concentrations (≥20 μM) it acts as a competitive substrate (both a substrate and an inhibitor). Because low concentrations of radiotracer are used in PET imaging, [11C]dLop will act only as a selective substrate for P-gp at the BBB.
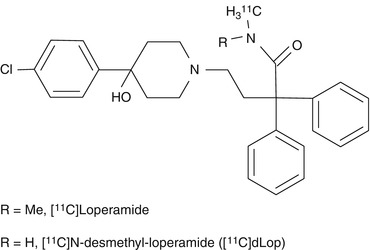
Fig. 21.2
Structures of [11C]loperamide and [11C]dLop
Pretreatment with the inhibitor tariquidar increases the brain uptake of [11C]dLop markedly in monkeys and humans (Kreisl et al. 2010). After entry of [11C]dLop into the brain, it is trapped and doesn’t wash out. Although dLop is an opiate agonist, its trapping is not a result of high-affinity binding to the opiate receptor. Brain uptake of [11C]dLop cannot be displaced by receptor-saturating doses of an opiate agonist or antagonist (Lazarova et al. 2008). This irreversible trapping amplifies the PET signal so it is advantageous from an imaging perspective. dLop has a pK a of 7.3 and may be trapped by passive diffusion into acidic organelles, mainly lysosomes, followed by protonation within the organelle (Kaufmann and Krise 2007; MacIntyre and Cutler 1988). To test this hypothesis the accumulation of [3H]dLop was measured in human cell lines and compared with the uptake of other weak bases (Kannan et al. 2011a). [11C]dLop was found to be ionically trapped in acidic lysosomes and tariquidar competed for lysosomal accumulation, both in vitro and in vivo. This interaction is problematic in peripheral organs but not in the brain. Thus [11C]dLop can still be used, either alone or in combination with tariquidar, to measure the function of P-gp at the BBB.
The low baseline uptake of radiolabeled substrates in the brain because of a very large capacity of pumping systems in the BBB is still an important issue, since it hampers the detection of subtle changes in function (particularly an upregulation of P-gp). Thus, novel PET ligands for P-gp with improved kinetics are still required. MC266 is a tracer of P-gp function with higher baseline uptake than [11C]verapamil (van Waarde et al. 2009). Thus, upregulation of P-gp function in response to treatment may be detectable using MC266 (Fig. 21.3) and PET. Another alternative is the P-gp inhibitor [11C]MC18 (Fig. 21.3), a unique tracer of P-gp expression which shows specific binding in the brain and several peripheral target organs. However, a derivative with higher affinity for P-gp may be required for quantitative imaging (van Waarde et al. 2009).
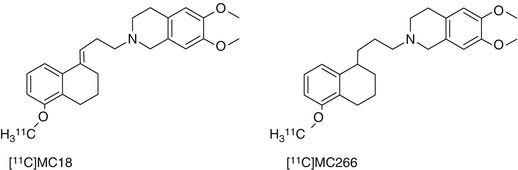
Fig. 21.3
Structures of [11C]MC18 and [11C]MC266
21.4.3 Inhibitors as Tracers for Measuring P-gp Expression
Because currently available PET tracers are high-affinity substrates for P-gp, they are efficiently kept out of the brain by P-gp-mediated transport and therefore show low brain uptake. Low brain uptake makes it difficult to use these probes for the mapping of regional differences in cerebral P-gp activity, which are expected to occur, for example, in medically refractory epilepsy. A promising complementary approach to the use of radiolabeled substrates for assessing P-gp in vivo would be the use of radiolabeled inhibitors, which bind with nanomolar affinity to the pump without being transported. Such tracers are expected to show a signal increase rather than a signal reduction in brain regions that overexpress P-gp.
21.4.3.1 [11C]Elacridar
[11C]Elacridar (Fig. 21.4) displayed high metabolic stability and displaceable binding to rat brain slices in vitro (Dörner et al. 2009). During subsequent in vivo studies, tracer concentration was about ten times higher in the brain of wild-type mice than in rat brain. This difference can most likely be explained by the severalfold higher blood activity levels in mice as compared to rats. [11C]Elacridar is a nontransported inhibitor of P-gp but also inhibits breast cancer resistance protein (BCRP, ABCG2) at comparable concentrations as it inhibits P-gp. However, elacridar does not inhibit multidrug resistance-associated proteins (MRPs) such as MRP1. Experiments in wild-type and mdr1a/b knockout mice showed comparable blood activity levels of the PET tracer, but in knockout animals brain uptake of [11C]elacridar was 2.5 times higher. Following administration of unlabeled elacridar, brain uptake of [11C]elacridar in rats was increased by a factor of 5 and in wild-type mice by a factor of 2.5. Similar results were obtained in rats and mice, but the optimal blood concentration (EC90) of elacridar in guinea pigs was tenfold higher, suggesting the existence of species differences in rodents (Cutler et al. 2006). Elacridar, as well as tariquidar (see below), was able to increase [11C]verapamil distribution in rat brain up to 11-fold over baseline at maximum effective doses and was about three times more potent than tariquidar (Kuntner et al. 2010), probably because of its dual action on both P-gp and BCRP. Further studies aimed at detecting P-gp and BCRP function were conducted by Dörner et al. who used 1-[18F]fluoroelacridar (Dörner et al. 2011). In conclusion, [11C]elacridar is not specific for P-gp and its signal magnitude is low.
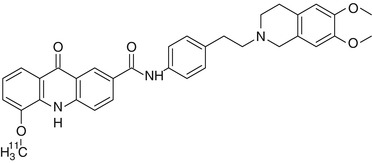
Fig. 21.4
Structure of [11C]elacridar
21.4.3.2 [11C]Tariquidar
Tariquidar is at low concentrations a noncompetitive inhibitor of P-gp and a substrate for breast cancer resistance protein (BCRP, ABCG2). At higher concentrations (≥100 nM) tariquidar acts as an inhibitor of both P-gp and BCRP (Kannan et al. 2011b; Kühnle et al. 2009). For this reason [11C]tariquidar is not a suitable PET tracer for mapping of cerebral BCRP expression (Mairinger et al. 2010). Tariquidar does not inhibit multidrug resistance-associated proteins (MRPs) such as MRP1 (Mistry et al. 2001). In the presence of unlabeled ligand, an increased influx of radioactivity into the brain has been observed (Bauer et al. 2010). Unlabeled tariquidar may have displaced labeled tariquidar from its binding site and may have resulted in passive diffusion of [11C]tariquidar (Fig. 21.5) into the brain. Advantages of [11C]tariquidar are the apparent lack of nonspecific binding to other membrane proteins and good metabolic stability. One hour after injection in rats, 96 % of plasma radioactivity represents unchanged parent compound (Bauer et al. 2010). Further experiments are needed to clarify if [11C]tariquidar behaves as a transported or as a nontransported P-gp/BCRP inhibitor in vivo. A new tariquidar-like compound has been synthesized as a BCRP-selective inhibitor (see Mairinger et al. 2010 for molecular structure). Regional differences in tariquidar-induced modulation of [11C]verapamil brain uptake indicate regional differences of P-gp function in rat brain (Kuntner et al. 2010). In addition, the use of 2 mg/kg body weight (BW) of tariquidar in rats demonstrated a P-gp inhibition comparable to that obtained in humans with 3 mg/kg (Kuntner et al. 2010). In contrast, other studies demonstrated that 6 mg/kg of tariquidar was the minimum dosage necessary to obtain a significant increase in the brain uptake of [11C]dLop (Kreisl et al. 2010). Noteworthy, Kurnik et al., who did not use PET, failed to demonstrate a significant effect of the 2 mg/kg dose of tariquidar on central opioid effects of loperamide in humans because of the greater resistance of brain P-gp to inhibition as compared to T lymphocytes (Kurnik et al. 2008). Tariquidar-induced P-gp modulation at the human BBB appears to be transient (depending on the species, tracers, and tissue), and its magnitude is directly proportional to serum drug exposure. Thus, higher plasma concentrations of tariquidar could be safely obtained in the clinic in order to overcome drug resistance and to achieve higher levels of cerebral P-gp inhibition (Wagner et al. 2009).
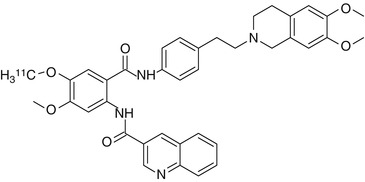
Fig. 21.5
Structure of [11C]tariquidar
21.4.3.3 [11C]Laniquidar
Laniquidar is a noncompetitive inhibitor which has been used in Phase I and II clinical trials (van Zuylen et al. 2000; 2002) as well as in animal studies (Bardelmeijer et al. 2004). Laniquidar lacks interactions with cytochrome P450 isoenzymes which should result in low levels of labeled metabolites (Thomas and Coley 2003). Lipophilicity of laniquidar is rather high (log P = 6.89) (Wang et al. 2003). A potential explanation of low cerebral uptake could be high binding to plasma proteins, resulting in a low free fraction of [11C]laniquidar (Fig. 21.6) in plasma. When [11C]laniquidar is injected in tracer amounts, it may act as a substrate rather than an inhibitor (Moerman et al. 2012). This is supported by pretreatment with cyclosporin A in rats which yielded in higher brain uptake of [11C]laniquidar (Luurtsema et al. 2009). Studies in WT mice demonstrate statistically higher brain uptake of [11C]laniquidar when administered with the cold compound as a carrier, representing a therapeutic dose of 60 mg/kg laniquidar (Moerman et al. 2012).
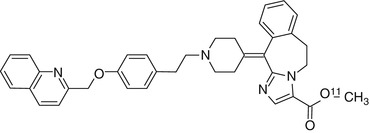
Fig. 21.6
Structure of [11C]laniquidar
It is important to mention that besides species differences, there are also some important factors that can influence P-gp expression/activity which must be taken into account. For example, estrogens may induce increases of P-gp function (see below). Thus, PET data obtained from female animals must be interpreted with care. In addition, isoflurane can cause an increase in neuroreceptor ligand binding although its effect on P-gp is unknown (Elfving et al. 2003; Harada et al. 2004).
21.4.4 PET Studies with [11C]Verapamil in Neurological Diseases
Kortekaas et al. showed that the intracerebral transfer of [11C]verapamil was significantly increased in the midbrain of patients with Parkinson’s disease (PD), when compared to healthy volunteers. Since verapamil is a P-gp substrate, this result supports a dysfunctional BBB as a correlated mechanism in this brain disease (Kortekaas et al. 2005). Bartels et al. found in ten advanced PD patients (5 ± 2 years runtime disease) treated with levodopa and dopaminergic agonists an increase of cerebral [11C]verapamil distribution in white matter of the frontal cortex, in comparison with healthy controls (Bartels et al. 2008a). On the other hand, ten young de novo PD patients presented a decrease of cerebral [11C]verapamil uptake in the brain stem, indicating a localized upregulation of P-gp at the onset of PD (Bartels et al. 2008a). So, the authors highlighted a delayed decrease of P-gp functionality, related with disease progression, and a similar change is observed even during normal aging. A decrease in P-gp activity at the blood–brain barrier level and in peripheral mononuclear blood cells was found in elderly healthy volunteers, in comparison to young healthy subjects (Bartels et al. 2008b; Toornvliet et al. 2006). In addition, a recent study using [11C]verapamil also found decreased P-gp function in patients with sporadic Alzheimer’s disease (van Assema et al. 2012b). However, this kind of studies should be interpreted with caution because of the possibility of drug substances, stress, and emotions affecting P-gp functionality. Using [11C]verapamil to assess BBB P-gp function in rats, de Klerk et al. showed that the antidepressant venlafaxine increases P-gp function whereas chronic stress inhibits it (de Klerk et al. 2010a). An interesting study revealed that the clearance of [11C]verapamil from the brain is significantly greater in younger monkeys than in adults. It was also shown that the brain penetration of [11C]oseltamivir changed with development and was slightly higher in younger monkeys (Takashima et al. 2011). The authors pointed out that this change may be related to the observed difference in drug responses in the brains of children and human adults. Taken together, these results suggest that besides disease, other factors can affect P-gp status. P-gp is a vulnerable protein; toxic substances can change its activity or expression depending on how much exposure is encountered during life.
The contribution of P-gp to regional drug distribution in healthy human brains was assessed using PET (Muzi et al. 2009; Sasongko et al. 2005). Fairly uniform P-gp activity was observed in all brain regions that are protected by the BBB and negligible activity in the pituitary, which is situated outside the BBB (Eyal et al. 2010).
Patients with chronic schizophrenia treated with antipsychotic drugs show a significant decrease in [11C]verapamil uptake in the temporal cortex, the basal ganglia, and the amygdala compared with healthy controls, and a trend towards a significant decrease is found throughout the brain. Such a decrease correlates with an increased activity of P-gp (de Klerk et al. 2010b), which may contribute to drug resistance in schizophrenia, induced by antipsychotics (Colabufo et al. 2010). So, because both pathology and medication can cause alterations in cell homeostasis and consequently in P-gp function and expression, drug-naïve patients should be examined besides medicated patients, although recruitment of such subjects is quite difficult.
21.4.5 Comparative Studies of [11C]Verapamil and Its Carbon-11-Labeled Metabolite D617 in BBB Using PET
Cerebral kinetics of the recently synthesized compound [11C]D617, a metabolite of [11C]verapamil, has been studied before and after administration of tariquidar. This study used Sprague–Dawley and Wistar rats in order to assess possible differences in tariquidar-induced effects between these two strains. The results showed an approximately twofold increase in the cerebral volume of distribution (V T) of [11C]D617 after intravascular administration of tariquidar in both rat strains. Results also indicated that the lack of a tariquidar-induced effect in the ex vivo biodistribution study was due to the route of administration. Comparison with previously reported [11C]verapamil data showed that, at baseline, the kinetics of [11C]D617 and (R)-[11C]verapamil are similar (Verbeek et al. 2012). It is important to note that a tenfold increase of [11C]verapamil uptake has been observed after treatment with the same dose of tariquidar, indicating that [11C]verapamil is a better P-gp substrate than [11C]D617. The similar kinetics of [11C]D617 and [11C]verapamil at baseline provides support for use of the kinetic model for analysis of [11C]verapamil data (Lubberink et al. 2007). Nevertheless, the authors argued that the different kinetics of [11C]D617 and [11C]verapamil after P-gp inhibition indicates that assumption about kinetics should be made with care, and further simulation studies are needed to assess the impact of such differences under different conditions (Verbeek et al. 2012). It is important to mention that a recent study already measured the test–retest (TRT) variability of regional [11C]verapamil uptake in addition to variability of total brain uptake (Lubberink et al. 2007). This work showed that reproducibility of [11C]verapamil PET studies was best if V T derived from single-tissue (global TRT 6 %) and constrained two-tissue (global TRT 9 %) compartment models was used as the outcome parameter. Thus, the two-tissue compartment model is the kinetic model of choice with the volume of distribution V T as the preferred outcome measure (van Assema et al. 2012a).
21.4.6 Comparative Studies of [11C]Verapamil and [11C]dLop in BBB Using PET
[11C]Verapamil generates more brain-penetrating radiometabolites than [11C]dLop in rats. Approximately 30 % of brain radioactivity reflects radiometabolites (Luurtsema et al. 2005), compared to less than 10 % after administration of [11C]dLop (Lazarova et al. 2008). In healthy human subjects, [11C]verapamil has a fourfold higher brain uptake than [11C]dLop (70 % vs. 15 % SUV – standardized uptake value, respectively), suggesting the generation of radiometabolites from [11C]verapamil (Ikoma et al. 2006; Seneca et al. 2009). In addition, the K 1 of [11C]verapamil (0.05 mLcm−3min−1) is about fivefold greater than that of [11C]dLop (0.009 mLcm−3min−1) (Seneca et al. 2009). [11C]dLop, as well as [11C]loperamide, is an avid substrate for P-gp (Zoghbi et al. 2008). In monkeys, the brain uptake of [11C]dLop is high (>40 %) when P-gp is inhibited and is dependent on regional blood flow (Liow et al. 2009). Such result was followed by the demonstration that P-gp function appears fairly uniformly distributed in the monkey brain (Liow et al. 2009). At baseline conditions, almost no [11C]dLop enters the brain because of the high capacity of P-gp. Thus, the baseline uptake of [11C]dLop is not sensitive to increases of P-gp function (Kannan et al. 2011a). However, it may be possible to use [11C]dLop in combination with nonradioactive tariquidar to detect regional increases of P-gp function after partial inhibition of the efflux pump. Extensive research is ongoing to check if this approach is feasible in human subjects. The study of pathological conditions with increased P-gp function (e.g., drug-resistant epilepsy) will also benefit from this approach. However, for brain disorders in which P-gp function may be reduced, such as Alzheimer’s disease (Vogelgesang et al. 2002, 2004), [11C]dLop may be advantageous because it may enter the brain and may provide a strong signal, as it is irreversibly trapped in lysosomes (Kannan et al. 2011a).
21.4.7 18F-Labeled Compounds to Assess P-gp Function and Expression
Several studies with 18F probes for P-gp have been reported. Such probes have a longer physical half-life in comparison to [11C]labeled compounds (110 min vs. 20 min) and can be transported to other PET sites which lack GMP (good manufacturing practice)-compliant radiopharmaceutical production facilities. The synthesis and in vivo evaluation of [18F]fluoroethyl elacridar and tariquidar as PET probes for assessing the function of drug efflux transporters P-gp and BCRP have been reported by Kawamura et al. Results of the two new 18F-labeled compounds were compared to those of the two [11C]labeled parent molecules that were tested in previous studies (Kawamura et al. 2010, 2011b). In addition, radioactivity levels in mice brain after injection of [18F]elacridar and [18F]tariquidar were increased when the function of drug efflux transporters was inhibited. The increase of the uptake of these two compounds was similar to that of their [11C]labeled analogs. In metabolite analyses of mice brain and plasma, [18F]tariquidar proved metabolically more stable than [18F]elacridar. The authors concluded that [18F]tariquidar may be more suitable than 18F-elacridar to evaluate the in vivo function of drug efflux transporters (Kawamura et al. 2011a), but that probe concentration needs to be chosen carefully in order to assess both P-gp and BCRP function (Kannan et al. 2011b).
21.4.8 Use of P-gp Stimulator Compounds as a Strategy for Treatment of Neurodegenerative Diseases
Recently a benzopyrane derivative was synthesized which demonstrated a potent P-gp ligand activity with increased selectivity for P-gp over the MRP1 pump when compared to the lead compound MC18 (Contino et al. 2012). However, initial results indicated that 1,2,3,4-tetrahydro-2-((3,4-dihydro-6-methoxy-2H-chromen-3-yl)-methyl)-6,7-dimethoxyisoquinoline is a P-gp-inducing agent, whereas MC18 is a potent P-gp inhibitor (Contino et al. 2012). Because of these findings the authors pointed to the potential application of P-gp stimulators in treating Alzheimer’s disease (AD) and Parkinson’s disease (PD), as such compounds could stimulate the clearance of β-amyloid or α-synuclein, respectively, in AD and PD (Contino et al. 2012). A noteworthy previous study using widely known compounds including rifampicin, dexamethasone, caffeine, verapamil, hyperforin, β-estradiol, and pentylenetetrazole already demonstrated the ability of these substances to improve the efflux of β-amyloid, which may slow down the progression of neurodegenerative disease (Abuznait et al. 2011a). Furthermore, a galloyl benzamide molecule has been successfully used to design new potent galloyl-based selective P-gp and MRP1 modulators (Pellicani et al. 2012).
21.5 Targeting ABC Transporter Regulation in the BBB: Considerations for Further Investigations with New Tracers and Treatment Outcomes
The notion that ABC transporters can be regulated by endogenous as well as exogenous factors is already well established. For example, focusing on endothelin receptor-1 (ET-1) signaling through the endothelin receptor type B (ETB), nitric oxide synthase (NOS), and protein kinase C (PKC) rapidly decreases P-gp activity in isolated rat brain capillaries (Hartz et al. 2004). Another report showed that BBB P-gp expression is regulated by the inflammatory mediators LPS, tumor necrosis factor-α (TNF-α), and ET-1, which activated toll-like receptor 4 (TLR4), TNF receptor 1 (TNFR1), ETB receptor, NOS, and PKC, leading to reduced P-gp activity (Hartz et al. 2006). Moreover, glutamate, cyclooxygenase-2, and prostaglandin E2-EP1 receptor signaling were implicated in seizure-induced upregulation of P-gp (Bankstahl et al. 2008a; Pekcec et al. 2009; Zibell et al. 2009). Not only P-gp but also other ABC transporters can be regulated by inflammatory mediators. Recently, it was shown that HIV-1 envelope glycoprotein gp120 and TNF-alpha are able to upregulate MRP1 expression in cultured astrocytes, and both NF-kappaB and c-Jun N-terminal kinase (JNK) signaling were involved in this regulation (Ronaldson et al. 2010). In addition, further studies demonstrated that BCRP mRNA levels and activity are significantly reduced by interleukin-1β (IL-1β), interleukin-6 (IL-6), and TNF-α (Poller et al. 2010). In a follow-up study, Rigor et al. identified PKC beta(I) as the responsible PKC isoform for downregulation of P-gp activity (Rigor et al. 2010). Importantly, this study demonstrated that targeting PKC beta(I) increases brain uptake of the P-gp substrate verapamil. Hawkins et al. made a similar observation using injection of vascular endothelial growth factor (VEGF). It was shown that VEGF decreased P-gp activity in rat brain capillaries via activation of VEGF receptors flk-1 and Src, probably through Src-mediated phosphorylation of caveolin-1 (Hawkins et al. 2010a). More studies are needed to determine whether targeting these pathways will improve the delivery of tracers across the BBB and into the brain.
Recently, sexual hormones have been implicated in efflux transporter modulation, especially estrogens (Sui et al. 2011). It was already demonstrated that estrogens play a key role in BCRP regulation (Imai et al. 2002). Estrone and 17β-estradiol reverse BCRP-mediated drug resistance, and the latter triggers posttranscriptional downregulation of BCRP in human breast cancer cell lines. Ee et al. identified an estrogen response element (ERE) in the BCRP promoter region and showed ERE activation by binding of the 17β-estradiol/estrogen receptor-α complex, which upregulated BCRP mRNA expression (Ee et al. 2004). In addition, Hartz et al. found that 17β-estradiol reduces BCRP transport activity in brain capillaries in a rapid way. Since this effect occurs within minutes and does not involve transcription, translation, or proteasomal degradation, a nongenomic mechanism is indicated (Hartz et al. 2010a). It is important to mention that P-gp activity is also upregulated by β-estradiol (Abuznait et al. 2011a, b). Thus, these results support the observation of gender-related differences in PET studies of P-gp function (van Assema et al. 2012c; Bebawy and Chetty 2009).
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Tracers for Beta-Amyloid and Other Proteinopathies
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 PET and SPECT Imaging of Steroid Hormone Receptors
PET and SPECT Imaging of Steroid Hormone Receptors
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


