Fig. 26.1
Chemical structures of clinically used sigma ligands for brain imaging
The first radioligand used for imaging sigma1 receptors in the CNS was [11C]SA4503 (Fig. 26.1), which was developed by Ishiwata and co-workers (2001). SA4503 has high and selective affinity for the sigma1 receptor (sigma1 IC50 = 17.4 nM, sigma2/sigma1 = 103) but low affinity for 36 neuroreceptors, ion channels and second messenger systems (Matsuno et al. 1996; Matsuno and Mita 1998). Later, three groups reinvestigated the binding properties of SA4503 and reported slightly different affinities and selectivities: sigma1 K i = 4.0–4.6 nM, sigma2/sigma1 = 13.3–55.0 (Lever et al. 2006; Shiba et al. 2006; Hirata et al. 2006). Density (B max) of the sigma receptor was estimated to be 30–600 fmol/mg protein (approximately 3–60 nM) in the human brain (Weissman et al. 1988; Kornhuber et al. 1996; Jansen et al. 1993). Theoretically, radioligands with nanomolar affinity (0.3–6 nM) will be suitable for sigma receptor imaging. From this viewpoint, SA4503 has a suitable range of affinity for quantitative in vivo imaging. In human studies, the use of [11C]SA4503-PEt allowed successful visualisation of the sigma1 receptor in the brain as described in Sect. 26.2.3.1, but the affinity of [11C]SA4503 was slightly high (i.e. the rate of dissociation of the ligand somewhat low) for measurement of the sigma1 receptor on the time scale of the PET scan using a 11C-labelled tracer with a short half-life (20.4 min) (Sakata et al. 2008).
The second PET ligand that was applied in human studies was [18F]FPS, which was developed by Waterhouse and co-workers (Collier et al. 1996). [18F]FPS was not cleared fast enough from the CNS to reach transient equilibrium by 4 h after administration in healthy volunteers due to its high affinity for the sigma1 receptor (K d = 0.5 nM) (Waterhouse et al. 2004; Zhao et al. 2005). In an effort to improve the CNS clearance for in vivo PET studies, Zhao et al. (2005) synthesised and evaluated [18F]SFE (previously designated as [18F]WLS1.002), a fluoroethyl derivative of [18F]FPS that exhibits a lower affinity for sigma1 receptors (K d = 5 nM). A preclinical study showed that [18F]SFE exhibits very similar regional brain distribution and specific binding in the rodent brain compared to [18F]FPS. As expected from its lower affinity, [18F]SFE is cleared much faster from the brain than [18F]FPS (Waterhouse et al. 2006a). Preclinical toxicity and dosimetry studies of [18F]SFE suggested that [18F]SFE will be safe for use in human PET studies (Waterhouse et al. 2006b). However, no further clinical imaging studies have been reported to date.
Carbon-11-labelled nemonapride, which was originally developed as a dopamine D2-like receptor radioligand (Hatano et al. 1989; Hatazawa et al. 1991), binds not only to D2-like receptors in the striatum but also to sigma receptors in other regions, such as the cerebral cortex and cerebellum (Ishiwata and Senda 1999), as has rather high affinities for both receptors. Nimura et al. (2004) applied [11C]nemonapride to imaging of sigma receptors in the cerebellum of patients suffering from levodopa-induced dyskinesia (LID), a brain area devoid of D2-like receptors.
More recently, two sigma1 receptor ligands with novel structures have been reported. Although these compounds showed promising properties for in vivo sigma1 receptor imaging, no clinical studies have been conducted to date. Maestrup and co-workers developed an innovative compound class of spirocyclic piperidines (Maestrup et al. 2009a, b). They found that a fluoroethyl derivative, [18F]fluspidine, had the best properties among the homologous fluoroalkyl-substituted spirocyclic sigma1 radiotracers (Maisonial et al. 2012). [18F]Fluspidine demonstrated favourable target affinity and specificity (sigma1 K i = 0.59 nM, sigma2/sigma1 = 1,330) as well as metabolic stability both in vitro and in animal experiments (Fischer et al. 2011). Moussa and co-workers developed a series of N-(2-benzofuranylmethyl)-N′-(alkoxybenzyl)piperazines as selective sigma1 receptor ligands. They found high affinity sigma1 selective ligands, such as the 4-[11C]methoxy (sigma1 K i = 2.7 nM, sigma2/sigma1 = 38) and 4-(2-[18F]fluoroethoxy) derivatives (sigma1 K i = 2.6, sigma2/sigma1 = 187) (Moussa et al. 2010; 2011). These compounds showed good brain uptake and specific binding to sigma1 receptors in the Papio hamadryas (baboon) brain. However, these compounds showed irreversible binding profiles during the scan periods because of their high affinities.
The sigma1 receptor-selective ligand [123I]TPCNE (K i = 0.67 nM, sigma2/sigma1 = 58) was used in a human SPECT study (Waterhouse et al. 1997; Stone et al. 2006). High brain uptake was reduced by haloperidol pretreatment, suggesting the specific binding of [123I]TPCNE to sigma1 receptors. However, the time-activity data were best described by an irreversible model. Thus, no further studies for this ligand have been conducted. Radioiodinated analogues of SA4503 were also prepared for SPECT, i.e. oI-SA4503 and mI-SA4503 (designated as o-BON and m-BON, respectively) (Hirata et al. 2006). The affinity and selectivity of mI-SA4503 for the sigma1 receptor (sigma1 K i = 8.9 nM, sigma2/sigma1 = 6.1) were approximately half those of SA4503. Although Hirata et al. (2008) used these compounds in a tumour study on rodents, no clinical studies have been conducted to date.
A review of the clinical trials for a few sigma1 receptor radioligands showed that ligands with affinities (K i) for the sigma1 receptor of 5–10 nM may be preferable for human studies and that [18F]FM-SA4503 may be more suitable than [11C]SA4503 (Kawamura et al. 2007). However, many sigma receptor ligands have affinity for the vesicular acetylcholine transporter (VAChT) (Efange 2000) and the EBP (Berardi et al. 2001). SA4503 was reported to show affinities for VAChT (K i = 50 nM, Shiba et al. 2006) and EBP (K i = 1.7 nM, Berardi et al. 2001), but [11C]SA4503 did not seem to bind to VAChT in the rat brain in vivo (Ishiwata et al. 2006a). Recently, Toyohara et al. (2012) confirmed that the brain uptake of [11C]SA4503 in mice was not blocked by high affinity EBP blockers tamoxifen (EBP K i = 2.8 nM, sigma1/EBP = 12) and trifluoperazine (EBP K i = 3.9 nM, sigam1/EBP = 52). The newly developed compound [18F]fluspidine showed weak affinity for VAChT (K i = 1,400 nM) and EBP (K i = 211 nM). Selectivity toward these binding sites should be taken into consideration when designing new selective ligands, although the relationship between the sigma receptor and EBP has not been clearly elucidated.
26.2.3 PET Imaging of the Sigma1 Receptors in the Human Brain
Three radioligands, [11C]SA4503 (Ishii et al. 2001; Sakata et al. 2007), [18F]FPS (Waterhouse et al. 2004) and [123I]TPCNE (Stone et al. 2006), were used for in vivo investigations of the density of the sigma1 receptor in the human brain by PET or SPECT; however, [18F]FPS and [123I]TPCNE showed irreversible binding profiles during the scan periods because of their high affinities. No further clinical studies were conducted. At present, clinical studies on sigma1 receptor imaging in living human brains are limited to [11C]SA4503 (Toyohara et al. 2009).
26.2.3.1 Healthy Subjects
[11C]SA4503 was distributed in all grey matter regions in the human brain (Fig. 26.2). The distribution patterns did not differ between the early and late phases and were similar to those of regional cerebral blood flow (rCBF). The regional tissue time-activity curves (TACs) of [11C]SA4503 in the brain showed a gradual increase in radioactivity over 30 min and then reached a plateau, whereas the radioactivity in plasma decreased very rapidly (Fig. 26.3). These findings suggest that the regional distributions of [11C]SA4503 could depend on the blood flow or permeability rather than receptor density because of its slow dissociation kinetics.
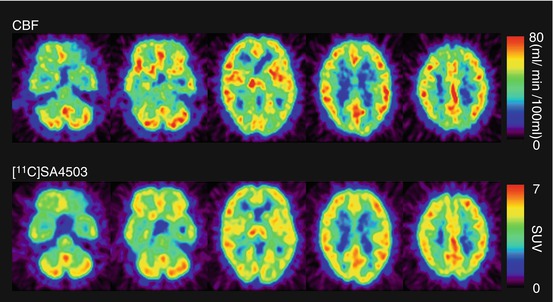
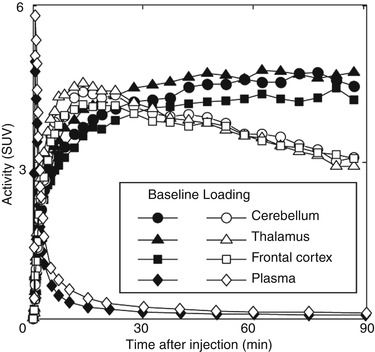
Fig. 26.2
rCBF and sigma1 receptors in the brain of a healthy human subject. rCBF (upper): rCBF was measured using PET and [15O]H2O, and calculated as ml/min/100 ml tissue. [11C]SA4503 (lower): Static images were acquired 40–60 min after injection of [11C]SA4503 and expressed as standardised uptake value (SUV: regional activity divided by administered dose per body weight)
Fig. 26.3
TACs in the healthy human brain and plasma after intravenous injection of [11C]SA4503 under baseline and haloperidol-loading (3 mg) conditions. Decay-corrected radioactivity was expressed as SUV. Haloperidol was administered orally 18 h before injection of [11C]SA4503
Preclinical studies confirmed age-dependent increases in sigma1 receptors in the brains of non-human primates (Kawamura et al. 2003). These results contrast strikingly with the age-dependent decreases in several other receptors in the primate brain. For investigation of the neuroprotective functions of sigma1 receptors, a PET study with [11C]SA4503 for evaluation of the aging process in the human brain would be of great interest.
26.2.3.2 Kinetic Analysis
In the traditional PET pharmacokinetics analysis for neuroreceptor radioligands, the ratio of k 3 (association constant rate between the free plus non-specifically and specifically bound compartments) over k 4 (dissociation constant rate between the free plus non-specifically and specifically bound compartments) in a two-tissue compartmental model (Mintun et al. 1984) is often used as the binding potential, which is currently defined as the specific binding potential relative to non-displaceable binding (BP ND) (Innis et al. 2007). As sigma1 receptors are distributed throughout all brain regions, no suitable reference region with negligible specific binding is available. Therefore, in the kinetic analysis of [11C]SA4503, arterial blood sampling measurements corrected by metabolite analysis are essential for obtaining the input function. In general, however, the variability of BP ND estimated from direct compartmental analysis without any constraints is clearly larger than that with indirect methods, such as a reference tissue model analysis (Vilkman et al. 2000). In the case of [11C]SA4503, slow kinetics due to its slightly higher affinity (Fig. 26.3) increased the variability of BP ND in the time scale of the PET scan using a 11C-labelled tracer with a short half-life (20.4 min). An alternative parameter for the evaluation of [11C]SA4503 binding is the total volume of distribution (V T) estimated by the linear graphical method (Logan et al. 1990), although V T includes both specific and non-specific binding (Kimura et al. 2007). The estimation of V T (K 1/k 2 × (1 + k 3/k 4)) in compartmental analysis tends to be more stable than that of BP ND. Figure 26.4 shows representative V T images of [11C]SA4503 in a healthy brain. The binding of [11C]SA4503 was high in the cerebellar cortex, moderate in the temporal and parietal cortices and low in the caudate and putamen (Sakata et al. 2007). These distribution patterns resemble the distribution patterns of radioligand binding in primate brains, where sigma receptors were noticed to be widely distributed with regionally different densities, using an in vitro binding assay and autoradiography (Weissman et al. 1988; Shibuya et al. 1992; Mash and Zabetian 1992).
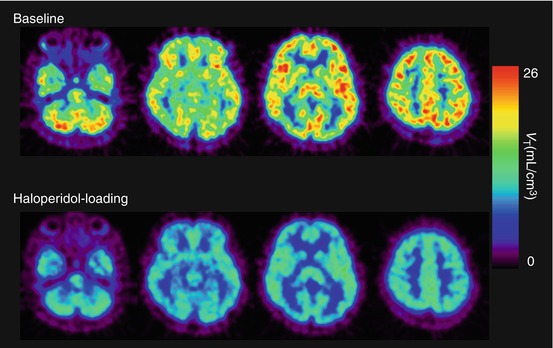
Fig. 26.4
V T images of [11C]SA4503 in the human brain under baseline (upper) and haloperidol-loading conditions (lower). The binding of [11C]SA4503 was considerably reduced after oral administration of haloperidol (3 mg, 18 h prior to the PET scan)
An interaction between sigma1 receptors and steroids has been established (Collier et al. 2007; Su et al. 1988). Currently, no information is available regarding the relationship between plasma hormone levels and [11C]SA4503 binding in the living human brain. If the binding of [11C]SA4503 is sensitive to competition by endogenous steroids, intra-subject variability in BP ND of [11C]SA4503 in women during the menstrual cycle and inter-subject variability in aged subjects, including post-menopausal subjects, may be found. Therefore, plasma steroid levels should be taken into account in PET studies of patients and healthy volunteers.
26.2.3.3 CNS Disease
Mishina et al. (2008) performed [11C]SA4503-PET in AD patients in the early stages of the disease. Figure 26.5 shows representative [11C]SA4503 images compared with those for glucose metabolism evaluated with [18F]FDG-PET. Although the results may have been influenced by atrophy or changes in regional blood flow, a widespread decrease in [11C]SA4503 binding in the brain was found; BP ND for [11C]SA4503 was significantly lower in the cerebellum, thalamus and frontal, temporal and occipital cortices of AD patients than in those of healthy subjects. In other regions of the AD brain, BP ND tended to be reduced. However, the data from these PET studies should be taken interpreted with caution because donepezil taken by some subjects shows a potent sigma1 receptor agonist activity (see Sects. 26.2.3.4 and 26.3.4).
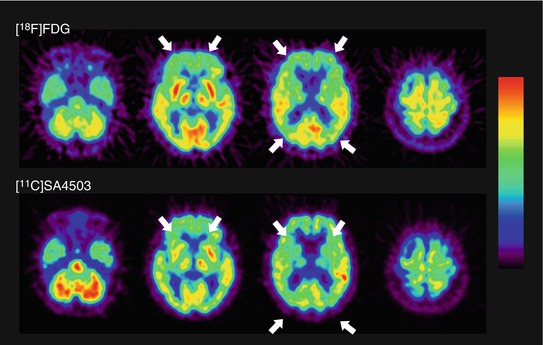
Fig. 26.5
Glucose metabolism and densities of sigma1 receptors in the brain of a patient with AD. Glucose metabolism (upper): static image acquired 45–51 min after injection of [18F]FDG. Sigma1 receptors (lower): V T derived from Logan graphical analysis of a 90-min dynamic scan of [11C]SA4503. In this patient, uptake of [18F]FDG was primarily decreased in the frontal and parietal cortex (arrow), and the distribution of [11C]SA4503 was consistent with this reduction (arrow)
Parkinson’s disease (PD) is also a progressive degenerative disorder of the CNS, characterised clinically by resting tremors, sluggish movements, cogwheel rigidity and postural instability. These symptoms are mainly caused by insufficient dopamine synthesis and death of dopaminergic neurons in the substantia nigra. Some studies have suggested that sigma1 receptors are involved in modulating the synthesis and release of dopamine (Chaki et al. 1998). Using [11C]SA4503-PET, Mishina et al. (2005) investigated whether sigma1 receptors were involved in the damage to the dopaminergic system in patients with PD, who showed low densities of dopamine transporters and normal or high densities of dopamine D2 receptors in the putamen using [11C]CFT and [11C]raclopride, respectively. Although the differences in BP ND of [11C]SA4503 between normal and PD patients were not clear because of a large inter-subject variability, BP ND in PD patients was significantly lower on the more affected side of the anterior putamen than on the less affected side (Fig. 26.6).
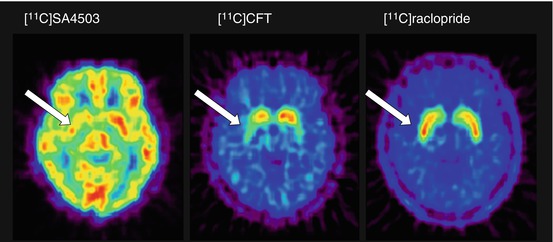
Fig. 26.6
Sigma1 receptors, dopamine transporters and dopamine D2 receptors in the brain of a patient with PD. [11C]SA4503 (left): V T derived from the Logan graphical analysis of a 90-min dynamic scan; [11C]CFT (middle) and [11C]raclopride (right): static image acquired 75–90 min and 40–55 min, respectively, after injection of the radiotracers. V T of [11C]SA4503 was smaller in the right anterior putamen (arrow). The uptake of [11C]CFT was reduced in the anterior putamen, especially on the right (arrow). The uptake of [11C]raclopride was preserved in the striatum and was larger in the right anterior putamen (arrow) than in the left
LID in patients with PD mimics acute dystonic reactions induced by antipsychotic drugs, possibly mediated by sigma receptors. A PET study using [11C]nemonapride showed a strong positive correlation between [11C]nemonapride binding and the preoperative LID severity score when the patients were receiving medication (Nimura et al. 2004).
26.2.3.4 Measurement of Sigma1 Receptor Occupancy in the Human Brain
While the physiological and pathophysiological roles of sigma receptors remain under investigation, a number of neuropsychiatric drugs are known to have moderate to high affinities for sigma receptors. Moreover, sigma1 receptor ligands represent a new class of therapeutic agents for neuropsychiatric disorders (Hashimoto and Ishiwata 2006; Ishikawa and Hashimoto 2010). Therefore, levels of sigma receptor occupancy of therapeutic drugs in the living human brain are of great interest. In a feasibility study, Ishiwata et al. (2006b) measured sigma1 receptor and dopamine D2 receptor occupancy by haloperidol in the human brain by PET using [11C]SA4503 and [11C]raclopride, respectively. Sigma1 receptor occupancy was approximately 80 % after oral administration of 3 mg haloperidol, whereas dopamine D2 receptor occupancy was approximately 60 %.
With respect to studies involving drugs with good potential for high receptor occupancy, a graphical analysis using a Lassen plot (Lassen et al. 1995; Cunningham et al. 2010) may be applicable for the evaluation of specific binding of the radioligand and receptor occupancy by the therapeutic drug, thus providing more stable estimates. The Lassen plot is based on regional changes in V T between baseline and drug-loaded conditions. The assumptions for the analysis are that non-specific binding is homogeneous, occupancy is the same in all regions of a regression line and there is a steady state of occupancy for the duration of the scan. A typical Lassen plot for haloperidol-loading study is shown in Fig. 26.7, and the parametric V T images of [11C]SA4503 for baseline and loading conditions (Fig. 26.4) show that the binding of [11C]SA4503 was globally decreased by haloperidol loading.
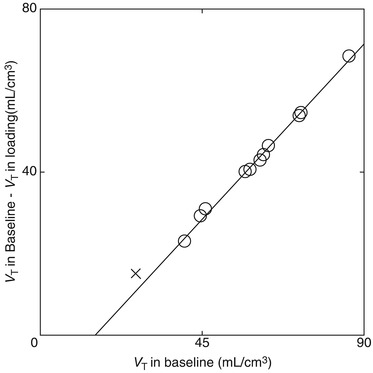
Fig. 26.7
Typical Lassen plot analysis applied to a haloperidol-loading study. In this plot, the x-axis is V T under baseline conditions and the y-axis is the difference between V T under loading and baseline conditions. The circles represent data from the grey matter regions (cerebral cortices, basal ganglia and cerebellar cortex) and the crosses represent data from the white matter region (centrum semiovale). The clear linear relationship suggests homogeneous non-specific binding across the grey matter regions investigated, and the x-intercept and gradient of the regression line (y = 0.96 (x – 15.1)) represent the volume of free plus non-specifically bound radioligand (V ND) and the receptor occupancy by non-radioactive haloperidol, respectively. Moreover, the absence of data near the x-axis indicates that no regions, including the white matter, are available as a true reference region devoid of specific binding, and that most [11C]SA4503 binding under baseline conditions is specific
Ishikawa et al. (2007) measured sigma1 receptor occupancy by two therapeutic drugs using [11C]SA4503-PET. Selective serotonin reuptake inhibitor (SSRI) is the treatment of choice for many disorders, including major depressive disorder, dysthymia, obsessive–compulsive disorder, and obsessive–compulsive spectrum disorders. Fluvoxamine has moderate affinity (K i = 36 nM) for sigma1 receptors in addition to the main affinity for serotonin reuptake sites (Narita et al. 1996). A single administration of therapeutic doses of fluvoxamine (50–200 mg) decreased [11C]SA4503 binding in the human brain in a dose-dependent manner, whereas that of paroxetine (20 mg), another SSRI with very low affinity for sigma1 receptors (K i = 1,893 nM), did not (Fig. 26.8). A similar occupancy study of an antipsychotic drug was also preliminarily described (van Waarde et al. 2011). The second example evaluated was donepezil. This drug has high affinity for sigma receptors (IC50 = 14 nM measured with [3H]DTG) (Kato et al. 1999). Figure 26.9 shows that a single administration of donepezil (5 or 10 mg) decreased [11C]SA4503 binding in the human brain in a dose-dependent manner (Ishikawa et al. 2009). The levels of sigma1 receptor occupancy by fluvoxamine and donepezil were approximately 60 and 75 %, respectively. These findings suggest that sigma1 receptors may be involved in the mechanism of action of fluvoxamine and donepezil.
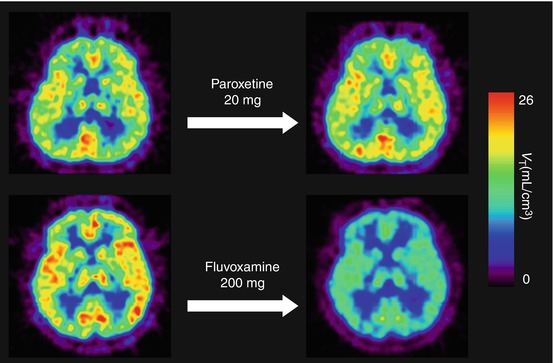
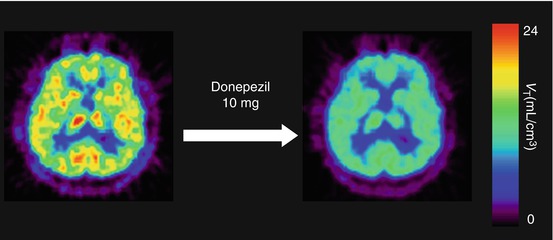
Fig. 26.8
V T images of [11C]SA4503-PET before and after a single oral administration of an SSRI. The upper pair represents V T images at baseline (left) and at paroxetine (20 mg) loading (right) in the same subject. The lower pair shows V T images at baseline (left) and fluvoxamine (200 mg) loading (right) in another subject
Fig. 26.9
Representative V T images of [11C]SA4503-PET before and after a single oral administration of donepezil (10 mg) in a healthy subject. Left image at baseline, right image after donepezil (10 mg) loading
26.3 Sigma Receptors in CNS Diseases
Since the prototypic sigma1 receptor ligand SKF10,047 (sigma1 K i = 44.8 nM, sigma2/sigma1 = 95.1; Walker et al. 1990) was found in the 1980s to exhibit psychotomimetic actions, the antipsychotic potential of sigma1 receptor ligands has been examined extensively. During the 1990s, sigma1 receptor ligands were synthesised mostly to develop new drugs for the treatment of schizophrenia. However, after cloning of the sigma1 receptor gene, molecular biological characterisation indicated relationships between sigma1 receptors and a variety of human diseases, including mood disorders, stroke, neurodegenerative diseases and drug addiction.
26.3.1 Schizophrenia
Five sigma1 receptor ligands, panamesine (EMD57445), eliprodil (SL82.0715), rimcazole (BW234U), BMY14802 (BMS181100) and DuP734, were introduced into clinical trials for treatment of schizophrenia (Hayashi and Su 2004). The findings of these clinical trials suggest that sigma1 receptor ligands may not possess potent antipsychotic actions against the positive symptoms of schizophrenia, but may be useful for ameliorating certain negative symptoms (Hayashi and Su 2004). Recently, it was reported that fluvoxamine, an SSRI possessing potent sigma1 receptor agonistic activity, improves negative symptoms and cognitive deficits of schizophrenic patients undergoing antipsychotic treatment (Iyo et al. 2008; Niitsu et al. 2010).
Using [3H]haloperidol (sigma1 K i = 0.3 nM, sigma2/sigma1 = 120; Walker et al 1990) in the presence of spiperone to block binding to dopamine D2 receptors, Weissman et al. (1991) first reported reductions in the density of sigma binding sites in schizophrenic patients on antipsychotic medication; these reductions were most prominent in the temporal cerebral cortex followed by the parietal cortex. Shibuya et al. (1992) measured sigma receptor binding with [3H]DTG (sigma1 K i = 11.9 nM, sigma2/sigma1 = 3.2; Walker et al. 1990) in 17 areas of the cerebral cortex and found no significant differences between schizophrenic patients and controls, with exception of the superior parietal cortex in which the binding was significantly increased in the schizophrenic group. In addition, no significant differences were observed between off-drug and on-drug schizophrenic patients in any brain areas. Helmeste et al. (1996) reported no differences in the binding of [3H]nemonapride (previously designated as [3H]YM-09151-2: sigma1 K i = 8.4 nM, sigma2/sigma1 = 1.1; Ujike et al. 1996) in the presence of spiperone in the frontal cortex and cerebellum; however, they observed a decrease in the caudate nucleus. The discrepancies between these results may have been due to differences in schizophrenia types, medication period, quantification methods (receptor binding assay or autoradiography) and radioligands used.
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Tracers for Beta-Amyloid and Other Proteinopathies
 PET Imaging of ABC Transporters in the BBB
PET Imaging of ABC Transporters in the BBB
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


