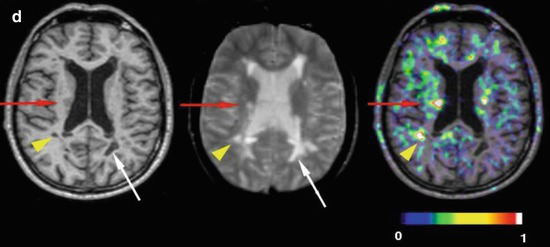
Fig. 22.1
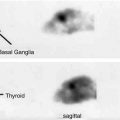
11C-PK11195 PET images of microglial activation in patients with (a) Alzheimer’s disease, (b) motor neurone disease, (c) Parkinson’s disease, (d) multiple sclerosis
11C-PK11195 and 11C-DAA1106 PET have also been reported to detect the presence of microglial activation in amnestic mild cognitive impairment (MCI). These subjects have isolated recall problems that are not severe enough to interfere with activities of daily living, but 60 % of these cases progress to develop Alzheimer’s disease (Petersen et al. 2001). In one series it was reported that 11C-PK11195 PET detected microglial activation in 40 % of amnestic MCI cases, while 60 % showed evidence of amyloid plaque deposition with 11C-PIB PET (Okello et al. 2009). However, others have failed to detect microglial activation in amnestic MCI with 11C-PK11195 PET (Schuitemaker et al. 2013). When a group of seven MCI cases were studied with 11C-DAA1106 PET, there was a significant mean of 27 % increase in tracer uptake across brain regions, and two cases individually had microglial activation elevated more than 2 SD above the control mean (Yasuno et al. 2012). Five of the seven MCI subjects with 11C-DAA1106 uptake raised more than 0.5 standard deviations above the normal mean progressed to frank dementia over a 2-year follow-up period.
22.4 Inflammation in Frontotemporal Dementia and Motor Neurone Disease
Dementia of the frontotemporal type affects around 10 % of dementia cases, and clinically it typically presents as personality change, praxis and language difficulties with memory becoming impaired later on. The pathology can involve Pick bodies containing three-repeat tau, TDP-43 protein inclusions or spongiform degeneration which targets the frontal and inferior temporal lobes. This dementia is not associated with amyloid plaque formation. A PET series has reported raised frontotemporal uptake of 11C-PK11195 in five FTLD cases (Cagnin et al. 2004).
There is both a clinical and pathological overlap between frontotemporal dementia and motor neurone disease (amyotrophic lateral sclerosis). The neuropathology of both can be associated with cortical TDP-43 inclusions and spongiform degeneration, and neuropsychological and imaging studies have indicated that dysfunction extends beyond the motor system in ALS. Ten probable or definite (El Escorial criteria) ALS patients and 14 healthy controls have been studied with 11C-PK11195 PET (Turner et al. 2004). Significantly increased microglial activation was seen in the motor cortex, brainstem, thalamus, dorsolateral prefrontal cortex and occipital lobes of the ALS patients – see Fig. 22.1b. There was a significant correlation between 11C-PK11195 uptake in the motor cortex and severity of upper motor neurone signs clinically.
22.5 Parkinsonian Syndromes and TSPO Imaging
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after dementia, affecting around 1 % of those over sixties. It manifests as asymmetrical limb bradykinesia, rigidity and tremor, and these symptoms are usually responsive to levodopa. The pathology is characterised by Lewy body inclusions in neurones which contain aggregated alpha-synuclein and neurofilaments.
While the dopamine neurones of the substantia nigra compacta are targeted by the Lewy body pathology, it is now thought that the pathology first affects the dorsal motor nucleus of the vagus in the medulla and then ascends through the brainstem to the cortex in stages (Braak et al. 2004). In stage 2 the locus coeruleus, pedunculopontine nucleus and median raphe in the pons become involved, while the dopamine neurones of the substantia nigra compacta in the midbrain only become targeted in stage 3 along with the cholinergic nucleus basalis. The limbic cortex and cingulate are involved by stage 4 and the association and primary neocortex in stages 5 and 6.
At postmortem microglial activation has been reported to accompany Lewy body pathology in both subcortical and cortical affected regions (Imamura et al. 2003; McGeer et al. 1993). It has been shown in vitro that aggregated alpha-synuclein fibrils can stimulate microglial activation, but the relationship between this inflammatory process and disease progression remains uncertain. In an initial PET study, it was reported that levels of increased midbrain 11C-PK11195 uptake in PD correlated with reductions in putamen dopamine transporter (DAT) binding measured with 11C-CFT PET (Ouchi et al. 2005). The authors suggested that this provided evidence for involvement of microglia in the loss of dopaminergic function that characterises PD. Later 11C-PK11195 PET series, however, have failed to detect consistent microglial activation in the substantia nigra of PD cases or to replicate an association between midbrain 11C-PK11195 signals and loss of dopaminergic function in PD. It is conceivable that the dopaminergic drugs used to treat PD may have an influence on the levels of microglial activation. Although none of these agents directly act to suppress microglial activation, it has been suggested that both dopamine agonists and monoamine oxidase B inhibitors have neuroprotective properties (Schapira and Olanow 2004). This claim is, however, controversial, and currently there is no hard evidence that therapy for PD influences levels of the inflammatory response.
Microglial activation is known to be widespread in PD, and raised 11C-PK11195 uptake has been reported in the brainstem, basal ganglia and cortical regions of non-demented cases, regions which are all targeted by Lewy body pathology in later disease stages – Fig. 22.1c (Gerhard et al. 2006a). We have recently found that levels of striatal 11C-PK11195 uptake in PD correlated with disability rated with the Unified Parkinson’s Disease Rating Scale (UPDRS), while impaired verbal fluency was associated with raised frontal and insular 11C-PK11195 activity (Simpson et al. 2012). This suggests that microglial activation may well reflect disease activity and is in line with the dying back theory of PD pathology which suggests that nigrostriatal terminals may become dysfunctional ahead of nigral cell body loss.
The level of 11C-PK11195 uptake appears to remain static over a 2-year follow-up period despite ongoing clinical deterioration and loss of putamen 18F-dopa storage of PD cases (Gerhard et al. 2006a). It has been suggested that microglial activation is an early phenomenon in PD and may act to drive disease progression and superadded dementia that can follow although levels of inflammation remain fixed. In favour of this viewpoint, it was noted that PD cases with late dementia had similar cortical levels of microglial activation to similarly disabled non-demented cases.
Neurodegenerative disorders that are associated with atypical parkinsonian disorders include multiple system atrophy (MSA) and progressive supranuclear palsy (PSP). MSA is associated with asymmetric parkinsonism along with early autonomic dysfunction, postural instability and ataxia. The pathology is characterised by argyrophilic glial cytoplasmic inclusions (GCIs) that contain alpha-synuclein and target the substantia nigra, putamen, pontocerebellar connections and the lateral columns of the spinal cord. Microglial activation is known to be associated with MSA pathology, and 11C-PK11195 PET has revealed raised binding in the putamen, pallidum, pons, substantia nigra and dorsolateral prefrontal cortex at higher levels than that associated with PD (Gerhard et al. 2003).
In a proteolipid protein-alpha-synuclein overexpression (PLP-αSYN) transgenic mouse model of MSA, a correlation between levels of nigral microglial activation and dopamine neurone loss has been reported (Stefanova et al. 2007). Minocycline administration was protective, both suppressing the inflammation and preserving dopamine neurones in the mouse. A human trial was subsequently performed to determine the neuroprotective efficacy of minocycline in MSA. The drug failed clinically to alter disease progression at clinically licensed doses; however, those MSA cases receiving active medication showed a 30 % reduction in brain 11C-PK11195 uptake relative to placebo-treated cases (Dodel et al. 2010).
Three PSP patients have been studied with 11C-PK11195 PET and reported to show significantly increased signal in their caudate nucleus, putamen, pallidum, midbrain, substantia nigra, frontal lobe and cerebellum (Gerhard et al. 2006b). One of these patients was rescanned after 10 months and their level of microglial activation had remained stable.
22.6 Huntington’s Disease and Microglial Activation
Huntington’s disease is an autosomal dominant inherited progressive neurodegenerative disorder associated with motor, cognitive and psychiatric symptoms. It is associated with an abnormal CAG triplet-repeat expansion of the huntingtin gene on chromosome 4 which leads to an elongated polyglutamine chain at the terminus of the huntingtin protein. The effect of this is to cause intranuclear aggregations of huntingtin to form resulting in progressive loss of medium spiny striatal GABAergic neurones (Sapp et al. 1997). These neurones express either dopamine D1 or D2 receptors, and so striatal dysfunction can be detected prior to symptom onset as a loss of availability of these sites for ligand binding (Andrews et al. 1999).
The loss of striatal neurones in HD is associated with microglial activation (Sapp et al. 2001) which can potentially be detected with 11C-PK11195 PET. Levels of striatal microglial activation correlate with loss of dopamine D2 receptors measured with 11C-raclopride PET and also with locomotor disability rated with the Unified Huntington’s Disease Rating Scale (UHDRS) (Pavese et al. 2006). This suggests that microglial activation may play a role in driving the disease process. In favour of this viewpoint, 11C-PK11195 PET studies have reported raised microglial activation in a majority of asymptomatic adult HD gene carriers (Tai et al. 2007). Those carriers with active disease also showed reduced dopamine D2 binding with 11C-raclopride PET. Cortical microglial activation can also be detected in pre-manifesting HD carriers confirming that this is not a purely basal ganglia disorder (Politis et al. 2011). Levels of striatal 11C-PK11195 uptake have been shown to correlate with the predicted time of clinical disease onset (Politis et al. 2011; Tai et al. 2007).
22.7 Measuring Inflammation in Multiple Sclerosis
Multiple sclerosis (MS) is a disease characterised clinically by relapsing and remitting neurological episodes followed by a progressive phase of disability. Pathologically central inflammatory demyelination is seen as white matter plaques associated with axonal degeneration. MS targets young adults and is the most common cause of non-traumatic disability in the Western world (Compston and Coles 2008). Active plaques of demyelination contain blood-borne B and T lymphocytes and macrophages that have invaded the disrupted blood-brain barrier. However, there is also an intrinsic immune reaction to the disease process manifested as involvement of activated microglia (Benveniste 2007). Postmortem investigations have detected activated microglia not just in white matter plaques but also in the cortex and apparently normal-appearing brain areas (De Groot et al. 2001; Peterson et al. 2001). In progressive MS activated microglia are seen surrounding degenerating myelinated axons (Magliozzi et al. 2010).
11C-PK11195 PET has been used to demonstrate the extent of microglial activation in MS patients – Fig. 22.1d (Banati et al. 2000; Debruyne et al. 2003). PET findings have been validated with 3H-PK11195 autoradiographic studies on human brain slices which showed that this tracer binds to activated microglia rather than astrocytes or lymphocytes in plaques of demyelination (Banati et al. 2000). PET reveals that not only can raised 11C-PK11195 uptake be seen in active plaques, where there is altered T2 signal and gadolinium enhancement on MRI, but also in normal-appearing white and grey matter (Banati et al. 2000; Debruyne et al. 2003; Versijpt et al. 2005). This finding supports the hypothesis that in early MS microglial activation may initiate the inflammatory process prior to invasion of the damaged blood-brain barrier by lymphocytes. The activated microglial load at baseline may well be the critical factor when predicting outcome in MS patients (Confavreux et al. 2000). 11C-PK11195 uptake has been reported to be higher in secondary progressive than relapsing-remitting MS. Additionally, a significant correlation was reported between levels of 11C-PK11195 binding in cortical grey matter and locomotor disability in patients with secondary progressive MS (Politis et al. 2012).
TSPO expression in MS has also been examined with 11C-PBR28 PET (Oh et al. 2010). Overall brain 11C-PBR28 uptake was increased in the MS cohort. Focal increases in binding, corresponding to areas of active inflammation and blood-brain barrier breakdown on gadolinium contrast-enhanced magnetic resonance imaging (MRI), were also evident. Increases in 11C-PBR28 binding preceded the appearance of contrast enhancement on magnetic resonance imaging in some lesions, suggesting that glial activation is an early phenomenon in MS lesion formation. Global levels of 11C-PBR28 binding correlated with disease duration but not with locomotor disability.
22.8 Traumatic Brain Injury
Recovery from concussive traumatic brain injury (TBI) is highly variable. With 11C-PK11195 PET it has been demonstrated that an inflammatory response is present in many of these cases years after their original injury. In a recent series ten patients had 11C-PK11195 PET at least 11 months after moderate to severe TBI, and binding was calculated in and around the site of focal brain damage and in selected distant and subcortical brain regions (Ramlackhansingh et al. 2011). Microglial activation was significantly raised in the thalami, putamen, occipital cortices, and posterior limb of the internal capsules after TBI though there was no increase in binding at the original site of the focal brain injury. Levels of thalamic 11C-PK11195 uptake were correlated with impairment on executive tasks but not with either the time since the injury or the amount of structural brain injury. The authors concluded that increased microglial activation can persist up to 17 years after TBI in distant brain areas from the original focal lesion, particularly in subcortical regions. They suggested that a chronic inflammatory response to TBI develops over time and that anti-microglial strategies may still be beneficial months to years after the original insult.
22.9 Stroke and Microglial Activation
Stroke is associated with microglial activation both around the infarct, which develops within hours of the event, but also later in disconnected brain regions including the thalamus and brainstem. In one series six patients were examined with 11C-PK11195 PET between 3 and 150 days after their infarct, and increased microglial activation was present in all the patients examined (Gerhard et al. 2005). In the first 6 days following the stroke, the PET lesions were generally smaller and adjacent to the MRI lesion with little spatial overlap. The size of the local area of microglial activation then increased over the next month overlapping with the MRI lesion as the blood-brain barrier became disrupted and invasion by macrophages binding 11C-PK11195 alongside local microglial activation occurred. By 6 months after the initial stroke, local microglial activation was subsiding, whereas distant areas of 11C-PK11195 uptake could be seen beyond the primary infarct site into connected areas of the same hemisphere, the thalamus and the brainstem. The cortical area with maximal 11C-PK11195 binding became atrophic during that 6-month interval.
22.10 Psychosis
Both dementia and Parkinson’s disease are associated with secondary psychosis related to cortical pathology and possibly the presence of cortical and limbic inflammation that can be detected in these disorders. Schizophrenia is a primary psychosis but has also been reported to be associated with low levels of brain inflammation at postmortem. In a recent series, seven schizophrenic patients in the recovery phase from a psychotic episode were studied with 11C-PK11195 PET (Doorduin et al. 2009). Conventional T1- and T2-weighted MRI had shown no significant abnormalities. PET revealed a 51 % increase in mean hippocampal and a 30 % increase in global cortical 11C-PK11195 binding in the schizophrenic subjects compared with age-matched cortical controls. Three of the subjects individually showed significantly raised levels of cortical inflammation. Previously, low-level inflammation in the cortex of schizophrenic cases in remission has been reported (van Berckel et al. 2008). The present authors suggested that active episodes of psychosis may be related to an increase in background inflammatory activity.
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 Progress in PET Imaging of the Norepinephrine Transporter System
Progress in PET Imaging of the Norepinephrine Transporter System
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 PET and SPECT Imaging of Steroid Hormone Receptors
PET and SPECT Imaging of Steroid Hormone Receptors
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


