Fig. 1.
The figure outlines the central steps of the photooxidation procedure. In the first row, the general scheme starts with the in vivo labeling of the cell cultures (endocytic uptake of the respective markers), followed by chemical fixation at defined uptake times. After the photooxidation step, with the conversion of the fluorescence signal into DAB-deposits, embedding and cutting of the different sections, these are analyzed in the LM and EM or processed for electron tomography and 3D-reconstruction. The middle part (second and third row) shows freshly fixed cultures in phase contrast compared to their fluorescence signals, which are taken before the onset of the illumination. Illumination then is accompanied on one hand by bleaching of the fluorescence, and on the other hand by precipitation of oxidized DAB. Ice crystals aside the objectives indicate necessary cooling during this step (see Note 18). The lower panel shows the final step of embedding of the cell cultures, with the coverslips now being put upside down on the resin. The Beem capsules should be slightly overfilled, in that the cell monolayers are well embedded within the resin. After polymerization, the coverslips are removed by repeated cycles of warming/cooling (touching the surface with liquid nitrogen and/or 60°C hot water). Afterwards, the cell monolayers are exposed at the surface of the resin ready for microtomy.
In this chapter, the method is illustrated through the analysis of the endocytic pathways of three molecules/particles, the lipid ceramide (Cer; Fig. 2), high density lipoproteins (HDL), and the plant lectin wheat germ agglutinin (WGA). BODIPY (BODIPY-Cer) and two Alexa dyes, Alexa568 (HDL-Alexa Fluor568) and Alexa555 (WGA-Alexa Fluor555), were used as fluorochromes. After addition to the cell culture medium, the binding, uptake, and intracellular progression of these molecules are identified by their fluorescent signals in the LM and after photooxidation by DAB-deposits in both the LM and EM.
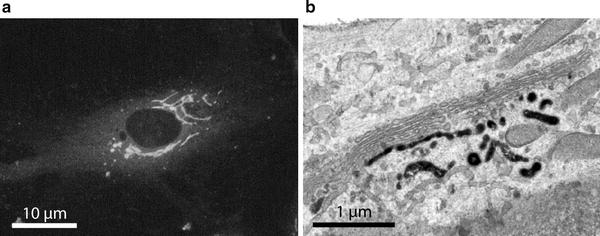
Fig. 2.
Localization of BODIPY-Cer in human endothelial cells. Cell culture was pre-incubated with BODIPY-Cer at 4°C for 15 min, warmed to 37°C, and incubated for 30 min. The lipid rapidly accumulated in the perinuclear Golgi region (a). After photooxidation, DAB deposits mark distinct Golgi regions: stacked Golgi cisternae as well as vesiculo-tubulo-cisternal membrane compartments of the trans-Golgi network (b).
2 Materials
2.1 Minor Equipment
General laboratory items such as gloves, work coats and masks, miscellaneous glass and plastic ware, adjustable pipettes, glass filtration device, filter paper, pH test strips, Beem capsules (polyethylene molds), Parafilm, and fine point tweezers for manipulation of the coverslips should be available.
2.2 Major Equipment
1.
Cell culture equipment.
2.
Inverse light microscope equipped with bright field and phase contrast and the respective filter combinations for fluorescence microscopy (FITC, TRITC).
3.
Transmission electron microscope including auxiliary infrastructure for fixation, embedding, polymerization, and ultramicrotomy.
4.
Fume hood.
5.
Ultrasonic bath.
6.
Magnetic stirrer.
2.3 Reagents
1.
BODIPY-ceramide (BODIPY®-FL-C5-ceramide conjugated to BSA).
2.
WGA-Alexa Fluor555.
3.
HDL-Alexa Fluor568.
5.
Sodium dihydrogen phosphate·2H2O (sodium phosphate monobasic dihydrate; NaH2PO4·2H2O).
6.
Di-sodium hydrogen phosphate·2H2O (sodium phosphate dibasic dihydrate; Na2HPO4·2H2O).
7.
Tris (hydroxymethyl) aminomethane (TRIS; 2-amino-2-(hydroxymethyl)-1,3-propanediol; H2NC(CH2OH)3); 0.05 M Tris–HCl buffer pH 7.4 (see Note 2).
8.
Cacodylic acid sodium salt (sodium cacodylate; dimethylarsinic acid sodium salt trihydrate (CH3)2AsO2Na . 3H2O; see Note 3).
10.
Sodium acetate (acetic acid sodium salt; CH3COONa).
16.
Potassium ferrocyanide (potassium hexacyanoferrate(II) trihydrate; K4[Fe(CN)6)·3H2O; see Note 10).
19.
Epon 812 (Glycid ether 100; 1,2,3-Propanetriol glycidyl ether); Epoxy resin of low viscosity (see Note 13).
20.
2-Dodecenylsuccinic acid anhydride (DDSA; C16H26O3); Epon hardener.
21.
Nadic methyl anhydride (NMA; methylnorbornene-2,3-dicarboxylic anhydride); Epon hardener.
22.
2,4,6-Tris(dimethylaminomethyl)phenol (DMP-30); accelerator for epoxy polymerization.
Alternatively: Epoxy Embedding Medium kit.
23.
Cells: Cultures of established cell lines are used in the experiments (HepG2 human hepatocarcinoma cells; WI-38 human fetal lung fibroblasts; Hs68 human foreskin fibroblasts; HUVEC human umbilical vein endothelial cells; all obtained from the American Type Culture Collection—ATCC).
2.4 General Processing of Cell Cultures
ATCC-human cell lines are cultured according to standard protocols (see Note 14). 24–48 h after seeding, they are in a subconfluent state being equally usable for LM and EM investigation. Model molecules/particles are added to the culture medium either at 4°C for pulse labeling or at 37°C for continuous uptake. For pulse labeling, coverslips with adhered cells are placed in Petri dishes with precooled medium for surface binding. After 15 min binding, labeled cultures are shortly rinsed in medium, replaced in Petri dishes with fresh medium, and incubated at 37°C for defined periods. Uptake and cellular progression of the molecules is stopped by rinses in cold buffer and immediate fixation.
The flowchart outlines the sequence of steps from the incubation of the cell cultures with fluorescent probes up to the preparation of sections for LM and EM. Take pictures for correlation of the experiment at these stages (see Note 19).
3 Methods
Wear gloves and protective coats throughout; pay attention to the safety instructions, especially when working with toxic, mutagenic, and/or biologic materials. Waste has to be collected according to the respective safety constraints/instructions.
3.1 BODIPY-Ceramide Photooxidation
2.
Cool down subconfluent cell cultures to 4°C for pulse labeling.
3.
Transfer coverslips with cell monolayers into fresh 3.5 cm diameter Petri dishes.
4.
Place 100 μL BODIPY-Cer solution on top of coverslips and incubate at 4°C for 15 min. Optional: Add Bodipy-Cer at 37°C for continuous uptake (see Note 16).
5.
Rinse several times with cold cell type-specific medium.
6.
Place coverslips in dishes with fresh medium.
7.
Incubate for indicated periods in culture medium at 37°C.
8.
Stop uptake of BODIPY-Cer by washing coverslips in cold PBS.
9.
Fix cells by placing the coverslips in Petri dishes with cold 4% FA + 0.5% GA in PBS at 4°C for 45 min (see Note 8).
10.
Parallel to the fixation of the samples, prepare the DAB solution, filter, and store it at 4°C in the dark (see Note 11).
11.
Wash cells in PBS.
12.
Place coverslips with labeled and fixed cells in fresh Petri dishes and wash again with Tris–HCl buffer.
13.
Pre-incubate with DAB/Tris–HCl buffer in the dark (cover with aluminum foil) for 15 min at room temperature.
14.
Replace DAB solution with 1 mL fresh DAB/Tris–HCl buffer and close the Petri dish.
15.
Put the closed Petri dish with the samples into a 10 cm diameter Petri dish and cover/surround with crashed ice.
16.
Put the Petri dishes together on an inverted light microscope and center the sample.
18.
Wash cells in 0.05 M Tris–HCl buffer pH 7.4.
19.
Wash cells in dH2O.
20.
Postfix with 1% osmium-ferrocyanide for 15 min. Osmium-ferrocyanide is freshly prepared before use by mixing aqueous 2% OsO4 and 3% potassium ferrocyanide 1:1 (see Notes 9 and 10).
21.
Dip coverslips into dH2O.
24.
Embed in Epon.
25.
Polymerize the resin.
3.2 WGA-Alexa Fluor555 Photooxidation
2.
Cool down subconfluent cell cultures to 4°C for pulse labeling.
3.
Transfer coverslips with cell layers into fresh 3.5 cm diameter Petri dishes.
4.
Place 100 μL WGA-Alexa Fluor555 solution on top of coverslips and incubate at 4°C for 15 min. Optional: Add WGA-Alexa Fluor555 at 37°C for continuous uptake (see Note 20).
5.
Rinse several times with cold cell type-specific medium.
6.
Place coverslips in dishes with fresh medium.
7.
Incubate for indicated periods in the cell culture medium at 37°C.
8.
 Histochemical Detection of Lipid Droplets in Cultured Cells
Histochemical Detection of Lipid Droplets in Cultured Cells
 Colocalization Analysis in Fluorescence Microscopy
High-Pressure Freezing for Scanning Transmission Electron Tomography Analysis of Cellular Organelles
Colocalization Analysis in Fluorescence Microscopy
High-Pressure Freezing for Scanning Transmission Electron Tomography Analysis of Cellular Organelles
 Morphological Analysis of Autophagy
Morphological Analysis of Autophagy
 A Novel Combined Imaging/Morphometrical Method for the Analysis of Human Sural Nerve Biopsies for Clinical Diagnosis
A Novel Combined Imaging/Morphometrical Method for the Analysis of Human Sural Nerve Biopsies for Clinical Diagnosis
 Environmental Scanning Electron Microscopy in Cell Biology
Environmental Scanning Electron Microscopy in Cell Biology




Stop uptake of WGA-Alexa Fluor555 by washing in cold PBS.
Related posts:
Histochemical Detection of Lipid Droplets in Cultured Cells
Colocalization Analysis in Fluorescence Microscopy
High-Pressure Freezing for Scanning Transmission Electron Tomography Analysis of Cellular Organelles
Morphological Analysis of Autophagy
Environmental Scanning Electron Microscopy in Cell Biology
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
