Fig. 1.
Photophysical states involved in photoswitching. (a) The structure and absorption spectra of mEosFPthermo before (left) and after (right ) activation. UV light induces an intramolecular reaction in which the protein backbone is cleaved and the π-system of the chromophore is extended. As a result, fluorescence absorption is red-shifted. (b) The structure and absorption of Alexa Fluor 532 in its active state (left ) and dark state (right ). Under intense illumination with excitation light, the fluorophore is driven into a triplet state and immediately reduced by thiols present in the imaging buffer. The formed radical shows no fluorescence and an increased absorption in the blue spectral range. Irradiation in the new absorption band drives a reaction with oxygen and allows the initial ground state of the fluorophore to be recovered.
Table 1
Photoswitchable fluorescent proteins
Name | Ex. Max. | Em. Max. | “On”/“off” contrast | Brightness (×103) | References |
|---|---|---|---|---|---|
PAGFP | 504 | 517 | 70 | 3 | (15) |
PSCFP2 | 490 | 511 | Poor | 11 | |
Dronpa | 503 | 522 | Good | 85 | |
BSDronpa | 460 | 504 | 17 | 22 | (25) |
Padron | 503 | 522 | 143 | 27 | (25) |
RSFastLime | 496 | 518 | 67 | 27 | (25) |
IrisFP (green) | 486 | 516 | 25 | (21) | |
PAmCherry1 | 564 | 595 | 4,000 | 8 | (32) |
PATagRFP | 562 | 595 | 540 | 25 | (33) |
Dendra2 | 553 | 573 | 19 | (29) | |
mKikGR | 580 | 591 | 33 | (30) | |
tdEosFP | 571 | 581 | 200 | 20 | (26) |
mEos2 | 573 | 584 | 30 | (23) | |
GFP* | 500–550 | 560 | (36) | ||
mIrisFP(red) | 546 | 578 | 19 | (21) | |
PSmOrange | 636 | 662 | 9 | (34) |
Many of the advantages of using fluorescent proteins are associated with their ability to be genetically fused to their targets (see Subheading 4.2). However, there are also drawbacks associated with this class of fluorophores. psFPs tend to emit less photons per “burst” than fluorescent dyes, and so often produce lower resolution images. “Off” state fluorescence is a reoccurring problem, and can be as high as 1% of “on” state fluorescence. Oligomerization is also a concern, as many psFPs are derived from parents that naturally exist as tetramers. Although protein engineers attempt to abolish this behavior, some residual tendency for aggregation can remain.
In this section, we review a selection of photoactivatable and photoswitchable fluorescent proteins. For a more detailed view on this rapidly growing field, we refer to excellent and recent reviews (18, 19).
2.1 Green Fluorescent Proteins
Although GFP itself has intrinsic photoswitching properties (20), the first effective photoswitchable fluorescent protein was paGFP (15). It is synthesized in a dark “off” state, but is irreversibly converted by irradiation with 405 nm light into a green-emitting, 488 nm excitable “on” state which can emit ∼160 detectable photons before bleaching (21). psCFP2 (22) is another fluorescent protein that emits in the same spectral window (green) as paGFP, but yields more detectable photons (∼250 median; (23)). In its “off” state, it emits blue photons when irradiated with 405 nm light; excitation of this blue form irreversibly converts the protein to a green-emitting “on” state excitable by 488 nm light. Unfortunately neither paGFP nor psCFP2 perform well in densely labeled samples; both have substantial “off” state fluorescence (∼1% of “on” state for paGFP; (15, 17)), and both are also activated to some extent by the 488 nm laser used to image their “on” states.
Several “reversible” green fluorescent proteins have also been developed. The first to be used for SMLM was Dronpa. This FP emits in the green channel, and has its excitation peak at 503 nm (24). Upon excitation, it emits a median of ∼120 detectable photons (17) before entering a stable dark state (with a half life of 840 min; (25)). UV illumination can return it to its “on” state. Dronpa can cycle many times before permanently photobleaching (24). It also appears to have a lower “off” state fluorescence than psCFP2 or paGFP, and so may be more appropriate for high density labeling. Several variants of Dronpa have been created that are slightly brighter or have different spectral properties; some have been used in SMLM experiments (25). One variant, Padron (25), has the opposite photophysical properties to Dronpa; it is switched “on,” and excited by 488 nm light, but switched “off” by 405 nm light. As Padron is not deactivated by excitation light, it may have a high photon yield.
IrisFP (21) is a complex photoactivatable FP with multiple switching behaviors. It initially emits in the green band under 488 nm excitation, but after releasing ∼170 detectable photons, switches to an inactive state. Exposure to 405 nm light drives the protein back into its original green-emitting state, while further UV exposure converts the green FP to an orange-emitting form excitable by a 568 nm light. This later form emits ∼350 detectable photons before photobleaching.
2.2 Orange Fluorescent Proteins
One of the first fluorescent proteins used in SMLM was the orange fluorescent protein EosFP (7). This protein is synthesized as a green emitting FP excitable by 488 nm, but can be—by exposure to UV light—converted to a orange-emitting “on” state excitable by 568 nm (26). The monomeric form of this protein matures poorly at 37°C, and thus seems unsuitable for expression in mammalian cells (26). However, its tandem dimer tdEos is fully functional at 37°C, and has been used extensively in SMLM, emitting a median ∼450 detectable photons before photobleaching (23).
An effort to improve EosFP led to the creation of mEos2, a variant that matures correctly at 37°C when expressed as a single fusion protein (i.e., not as a tandem dimer). It is now widely used in SMLM, and has an excellent photon yield (median ∼360 detected; (23)). Unfortunately, mEos2 has several properties that can cause problems in SMLM experiments. It has a tendency to aggregate in vitro, and thus may cause artifactual clustering of its fusion partner (23). Activated mEos2 molecules have also been shown to enter long-lived dark states before photobleaching (27, 28); thus in a typical SMLM experiment, individual molecules can be localized several times. For quantitative studies of protein clustering, careful control experiments are needed, as bona fide molecular clusters cannot be easily distinguished from single mEos2 molecules that have been localized multiple times. Similar blinking behavior has also been reported for its EosFP parent (26). Dendra2 and mKikGR are both monomeric green-to-orange photocovertable FPs that lack the tendency of mEos2 to aggregate (29, 30); however, neither yields as many photons as mEos2 (e.g., ∼130 median detected for Dendra2; (23, 31)).
Several dark-to-orange photoactivatable FPs have also been developed. PAmCherry1 is reported to yield as many photons as mEosFP (median 413 detected under 568 nm excitation; (32)) and has virtually no “off” state fluorescence. PATagRFP, a second dark-to-red FP, is three times brighter, and roughly twice as photostable as PAmCherry1 (33) in ensemble measurements, and so should also be useful in SMLM experiments. As the fluorescence emission of PATagRFP and PAmCherry1 are spectrally distinct from those of the green fluorescent proteins noted above, they can be used in two-color experiments.
2.3 Red Fluorescent Proteins
Recently, a new orange-to-red photoswitchable FP, PSmOrange, has been introduced. This psFP is revolutionary as it is the first to possess a red-emitting “on” state. It is initially orange, with peak emission at 565, but, upon exposure to 488 nm light, switches to a red-emitting FP with excitation and emission spectra comparable to those of Cy5 (34). Unfortunately, the photophysical properties of this psFP do not allow it to be localized as accurately as other psFPs (e.g., PAmCherry1). However, its red excitation/emission spectra may make it useful for in vivo experiments.
2.4 Conventional Fluorescent Proteins
In addition to fluorescent proteins that have been designed specifically for photoswitching, a number of conventional FPs have been used as photoswitches. GFP, for example, has long been known to enter stable nonfluorescent off states under constant 488 nm excitation; molecules can be returned to their active states by irradiation with UV light (20). EYFP can be similarly operated as a photoswitch, and has been used for super-resolution imaging in bacteria (35). A different strategy has been presented by the Sedat lab who used “oxidative reddening” to operate EGFP as a photoswitch, and demonstrated super-resolution imaging of histone proteins (36).
2.5 Preserving Fluorescent Protein Activity
Although the fluorophore inside a fluorescent protein is somewhat isolated from their environment by the beta barrel surrounding it, their photophysical properties can be easily altered, or destroyed, by exposure to buffers components. Chemical fixation, for example, can inactivate or reduce the activity of psFPs. We and others have found that glutaraldehyde is particularly destructive (18), while formaldehyde has more moderate effects (e.g., formaldehyde does not affect GFP fluorescence; (37)). Other data suggests that fixation can—in some cases—reduce, but not destroy the photon yield of psFPs (32). Mounting media might also have effects on the switching of psFPs. Such media are used to match the refractive index of a sample to that of the coverslip, in order to avoid imaging aberrations. However, most SMLM studies to date have not used mounting media but rather, immersed the samples in innocuous aqueous buffers (e.g., PBS or PHEM; (17, 32, 38)), as a result, little is known about the effects of mounting media on psFP activity. However, one can speculate that the redox environments and reduced oxygen levels of some mounting media might have strong effects on psFP activity. A recent study, for example, has found that reducing agents and oxygen depletion can both influence psFP behavior (28) (the influence of redox reagents is further discussed in Subheadings 5.1 and 5.2.4). However, we do note that recent SMLM studies that did not use special mounting media have achieved excellent results, probably because they observed only molecules found relatively close to the coverslip (using total internal reflection microscopy, which requires refractive index mismatch). As SMLM is applied to molecules found deeper in samples, we imagine that the issue of mounting media compatibility will become increasingly important.
3 Photoswitchable Fluorescent Dyes
Although psFPs are powerful tools for SMLM, it is sometimes desirable to use fluorescent dyes instead. These fluorophores exhibit photon yields that are considerably higher than those of fluorescent proteins, a fact that directly translates to increased localization accuracy and spatial resolution. Under standard imaging conditions (e.g., phosphate buffer saline), synthetic dyes typically spend most of their time in an “on” state. However, several different strategies have been developed that allow dyes to be switched “off,” most of which involve redox chemical processes. Novel dyes, which can be activated by the light-induced cleavage of a quenching group (39) have also been developed, and thus can be operated similarly to irreversibly activatable FPs.
3.1 Conventional Fluorophores
Many standard, commercially available dyes can be operated as photoswitches when imaged in buffers that allow excited fluorophores to stochastically enter reversible but long-lived dark states (e.g., containing oxygen scavengers and/or reducing agents). Under these conditions, fluorophores continuously exposed to a moderate intensity of excitation light begin in an “on” state, but transit quickly into a stable “off” state. Individual molecules exit their “off” states stochastically (induced by e.g., “activation” light or oxygen), and emit a burst of photons before being once again driven into an “off” state. The fraction of molecules in their “off” state is determined by the excitation laser intensity and buffer conditions; both should be optimized such that single fluorophores are distinguishable.
One bright and reliable photoswitch is Cy5 (or its derivative, Alexa Fluor 647; Fig. 2). When imaged (using 647 nm excitation) in buffers containing the reducing agent β-mercaptoethylamine (MEA) and lacking oxygen, it emits several thousand detectable photons before entering a long-lived dark state (τ 1/2 = 23 min; (40)). Inactive molecules can be stochastically and controllably brought back into their active states by irradiation with short wavelength light (405 nm, 488 nm, and 532 nm laser lines are all adequate). The structurally similar carbocyanines Cy5.5, Cy7 and Alexa Fluor 700 exhibit similar photoswitching properties (41).
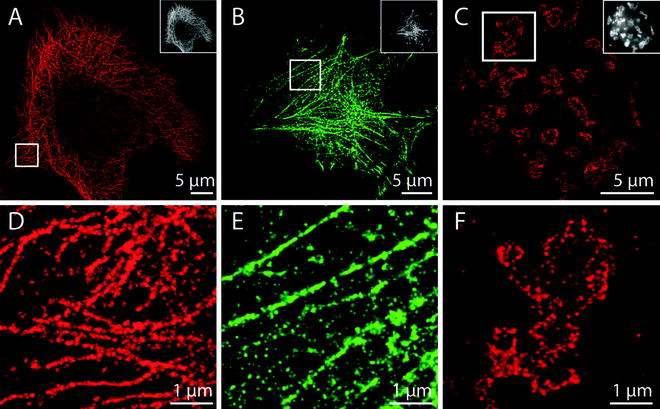
Fig. 2.
SMLM imaging of fixed HeLa cells. (a) Alpha-tubulin was immunostained with Alexa Fluor 647 and then imaged in PBS + 50 mM MEA (pH 7.4) using 647 nm excitation light (20,000 frames). Fluorophores in the dark state were controllably reactivated using 405 nm light. (b) Cells transfected with actin-mEos2 plasmids were imaged with an excitation wavelength of 568 nm and an activation wavelength of 405 nm (8,000 frames). (c) Giantin was immunostained with Alexa Fluor 647 and imaged in the presence of 100 mM MEA (pH 7.4) and an oxygen scavenger (6,000 frames) using 647 nm excitation. (d), (e), and (f) show magnified sections of the images in (a), (b), and (c).
A wide variety of other conventional fluorescent dyes, ranging from the green dye Alexa Fluor 488 to the far red dye ATTO 700, can also be used as reversible photoswitches in the presence of varying concentrations of MEA (Fig. 2; (42)). Such approaches are straightforward, but require freshly prepared MEA solution as well as careful control of buffer pH.
The mechanism through which reducing thiols create dark states is, for many fluorophores, now well understood. In the case of Rhodamine fluorophores such as Alexa Fluor 488, the nonfluorescent state that is generated has been identified as a radical anion using both absorption spectra and electron paramagnetic resonance (EPR) spectroscopy (Fig. 1b; (43)). These radical states have a lifetime of minutes in aqueous buffers, and are thus well suited for stochastic super-resolution imaging of dense samples (where only a small subset of all fluorophores present can be in the fluorescent state; (16)). Oxazine fluorophores, such as ATTO655, exhibit only very weak EPR signals (43); however, Fourier transform infrared (FT-IR) spectroscopy and theoretical calculations have demonstrated that the photoswitched nonfluorescent state is exhibited by fully reduced molecules (44). Photoswitching can often be controlled by adding both reducing and oxidizing reagents to buffers, as in the reducing and oxidizing system (ROXS) approach (45). ROXS allows engineering dark states by adjusting the concentrations of the buffer components that make SMLM possible (14). A number of similar approaches that rely on “on”/“off” switching of conventional synthetic fluorophores through different photophysical routes have been published in the recent years (46–48).
3.2 Activator–Reporter Dye Pairs
Pairs of fluorophores held in close spatial proximity can form systems with unique photoswitching properties (49). These photosystems consist of a “reporter” dye (e.g., Cy5), which is imaged directly, and an “activator” dye (e.g., Cy3) placed nearby. When the reporter dye is excited in a buffer containing a reducing agent and lacking oxygen, it quickly enters a dark state (as with direct switching), but can be reactivated by exciting the “activator” dye.
Replacing the “activator” and “reporter” with different dyes (e.g., replacing Cy5 with Cy5.5 or Cy7) yields probes that emit, or are activated, at different wavelengths (41). Such photoswitches are particularly useful for multicolor imaging. Although activator–reporter pairs are a proven tool for SMLM, they have the one drawback that they are not commercially available and so must be synthesized by the user.
3.3 Caged Fluorophores
Caged fluorophores are nonfluorescent in their original state and are activated by irradiation with light. Activation involves a chemical bond breakage (often, internal esters or lactones are used) which modifies the chemical structure of the synthetic fluorophore (e.g., establishes a conjugated π-system), and causes the probes to become fluorescent. Some caged fluorophores have been optimized for super-resolution imaging (39, 50). This strategy allows the number of fluorophores active in a sample to be finely controlled by modulating the intensity of activation light (much like irreversibly activatable psFPs).
In general, SMLM can be performed using any fluorophore that can be driven into a dark state, and a number of approaches have been developed in the past years which are beyond the scope of this article. The average time that the fluorophore resides in this dark state, however, has to be long enough to generate a single-molecule density at the sample (16). Noticeably, an early experimental demonstration of super-resolution imaging through stochastic switching used quantum dots as fluorophores, which exhibit an intrinsic blinking (51).
4 Labeling Strategies
4.1 Antibodies
One of the easiest ways to label a biomolecule of interest with photoswitchable fluorophores is to use an antibody. This strategy (i.e., immunohistochemistry) has long been a standard method in molecular biology, and has been described in detail elsewhere (52). Typically, cells are fixed, permeabilized, incubated with an antibody that binds the protein of interest (i.e., the primary antibody), and then incubated with a second, fluorescently labeled antibody which binds to the first.
This labeling strategy has multiple advantages. It can be performed on any fixed sample (including primary tissue), and does not require genetic manipulation of target cells. Samples can, theoretically, be labeled and imaged in a single day, although optimization of the labeling procedures is often required in practice. Importantly, labeling can be accomplished using commercially available primary and fluorophore-conjugated secondary antibodies.
However, the technique also has substantial drawbacks. It is difficult, for example, to prove that a primary antibody is binding specifically to its intended target, and not nonspecifically to other antigens, or “sticky” cellular structures. It is also nontrivial to determine what fraction of the target protein is bound; antigens can go unlabeled because they are sterically inaccessible to antibodies, or because they are unrecognizably denatured by fixatives (53). Both problems can be reduced, but not eliminated, by using “light” fixation procedures (e.g., a low concentration of formaldehyde).
Another notable disadvantage of traditional immunohistochemistry is that the complex formed by primary and secondary antibodies leaves fluorophores ∼10–20 nm away from their target (Fig. 3). As a result, the resolution achievable using this method will be >20 nm even if individual fluorophores that mark a target can be localized to a much higher accuracy. This problem can be lessened by labeling primary antibodies directly with fluorophores (using e.g., NHS-esters), or by isolating and labeling only the part of the primary antibody that binds the antigen (i.e., the Fab). The use of commercial antibodies heavily labeled with fluorophores can also create problems. When two fluorophores are within close proximity of each other, their photophysical properties can change dramatically. As a result, fluorophores bound to highly labeled antibodies can often fail to switch.
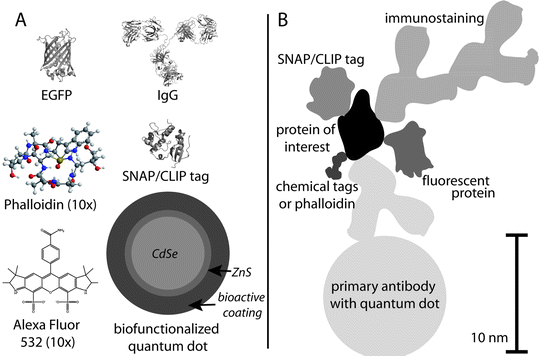
Fig. 3.
An overview of the tools currently available for the labeling of biomolecules with photoswitchable fluorophores. (a) Comparison of the size of different molecules used for fluorescence labeling. (b) An image of a hypothetical protein labeled using different strategies. Antibodies leave their fluorophores ∼15 nm away from their target, while fluorescent proteins and SNAP tags bring their fluorophores to within 1–2 nm.
4.2 Genetic Tags
Another widely used strategy for labeling a protein of interest with a photoswitchable fluorophore is to alter the target’s genetic code so that it is fused (through its N- or C terminus) to a genetically encoded tag. Such tags are usually fluorescent proteins, but can also be “chemical tags,” which bind small fluorescent molecules added to the surrounding media.
Although the genetic tagging of a protein is a relatively straightforward process, it can also be time-consuming. In bacteria and yeast, tagging can usually be accomplished by directly altering the genomic DNA of the organism and effectively replacing the endogenously expressed protein (54). In mammalian cells, plasmids expressing the tagged protein can be introduced transiently into cells using various transfection methods. However, more reliable biological results are often obtained when “stable” transfectants are selected (55); such cells have incorporated the exogenous DNA directly into their chromosomes, and thus do not have to be exposed to toxic transfection reagents directly before being imaged. Unfortunately mammalian cells usually express endogenous, unlabeled versions of target proteins. This problem can be solved by simultaneously knocking down the native protein with RNAi (targeting 3′UTRs), or by tagging the endogenous protein directly using zinc-finger nucleases.
Several crucial controls must be performed when using genetically encoded tags. As the presence of a bulky tag can reduce or eliminate the activity of a protein of interest, fusion proteins must be shown to be functional. One must also ensure that the tag being visualized in the cell is indeed bound to its target using a western blot (as cleavage at linker regions is not uncommon). Such blots can also reveal the degree to which a protein is overexpressed. Excess amounts of a target protein can cause mislocalization/aggregation in addition to adverse physiological effects.
In spite of these difficulties, genetic tags remain an extremely effective way of localizing cellular proteins. If used correctly, they can allow for the even labeling and visualization of most, if not all, molecules of a given protein. They are also able to position fluorophores very close to target molecules, a feature that should become increasingly important as SMLM-based methods mature (Fig. 3). In addition, they allow for the tagging of proteins for which no effective antibody exists.
Related posts:
 Introduction: Nanoimaging Techniques in Biology
Introduction: Nanoimaging Techniques in Biology
 Two-Color STED Imaging of Synapses in Living Brain Slices
Two-Color STED Imaging of Synapses in Living Brain Slices
 Secondary Ion Mass Spectrometry Imaging of Biological Membranes at High Spatial Resolution
Secondary Ion Mass Spectrometry Imaging of Biological Membranes at High Spatial Resolution
 Functional AFM Imaging of Cellular Membranes Using Functionalized Tips
Functional AFM Imaging of Cellular Membranes Using Functionalized Tips
 Correlative Optical and Scanning Probe Microscopies for Mapping Interactions at Membranes
Correlative Optical and Scanning Probe Microscopies for Mapping Interactions at Membranes
 Nanoimaging Cells Using Soft X-Ray Tomography
Nanoimaging Cells Using Soft X-Ray Tomography
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


