Etiology
Immune-mediated muscle inflammation and vascular damage are the hallmarks of polymyositis (PM) and dermatomyositis (DM). In PM the immune system is primed to act against muscle antigens, whereas in DM there is complement-mediated damage to both endomysial vessels and the microvasculature of the dermis. The trigger for the autoimmune malfunction in PM/DM is unknown. As with other connective tissue diseases, B-cell and T-cell autoregulation is disrupted. Viruses have a possible etiologic role, but direct evidence is lacking. However, a seasonal pattern of development of the disorders provides some epidemiologic evidence for an infectious agent having a causative role in PM and DM. In patients with anti–Jo-1 antibodies, weakness is more likely to develop in April, and those with a different autoantibody (anti–signal recognition particle) are more likely to have an onset of weakness in November. Animal models have been used to provide supportive evidence for virally induced myositis, and many different infectious agents have been proposed, including coxsackievirus, parvovirus, enterovirus, human T-lymphotropic virus, and human immunodeficiency virus.
Prevalence and Epidemiology
PM/DM has an incidence of approximately 5 to 10 cases per million per year, and in DM there are two population age peaks: children and young to middle-aged adults. PM affects women more frequently than men (2 : 1 ratio) and has a higher incidence in African Americans than in whites. In PM/DM pulmonary disease can overshadow the primary muscle disorder, and patients with PM/DM and interstitial lung disease (ILD) have higher mortality than those with isolated ILD. Estimates of PM/DM-related death rates vary between 10% and 27%, and these deaths are usually the consequence of cancer or pulmonary complications. Predictors of poor outcome include old age at onset, male gender, dysphagia, long-standing symptoms before diagnosis or therapy, pulmonary and cardiac involvement, and the presence of anti–Jo-1 antibodies. The majority of survivors have a chronic continuous or polycyclic disease course (80%).
Clinical Presentation
PM and DM are usually considered part of the same disease spectrum, and the main clinical difference is thought to be the prominent lilac “heliotrope” facial rash seen in DM. Nevertheless, DM and PM are pathogenetically distinct. DM is a humorally mediated microangiopathy, whereas PM is a cell-mediated immune response to muscle fibers. However, PM and DM are usually grouped together in many clinical studies, and for the purposes of this discussion they will be frequently considered a single entity.
The onset of PM/DM may be acute or insidious. Acute infection can precede or perhaps incite symptoms; children with juvenile DM frequently have symptoms of systemic illness 3 months before the onset of disease. DM (but not PM) is seen in the pediatric population, and symptoms are similar in children and adults, although childhood onset is more likely to be acute and florid.
The clinical hallmark of PM is proximal muscle weakness with muscle tenderness and myalgia. Patients may also have dyspnea, dysphagia, aspiration pneumonia, fever, weight loss, secondary Sjögren syndrome, Raynaud phenomenon, polyarthritis, and cardiac myopathy. The frequency of ILD is not definitively known in PM/DM patients; however, depending on inclusion criteria and follow-up length, upwards of 75% of patients will have some evidence of ILD. With the use of chest radiography alone, the incidence of ILD in patients with PM/DM has been estimated to be much lower at 9% to 10%, but this likely reflects the lesser sensitivity of radiography than high-resolution CT (HRCT). The association between polyarthritis and ILD in PM/DM is well known, and the pulmonary manifestations of PM/DM are thought to precede symptoms of the inflammatory myopathy in almost a third of cases. There are various modes of pulmonary involvement, ranging from fulminant and acute (diffuse alveolar damage, pneumonia) to chronic progressive symptoms (fibrotic lung disease, pulmonary hypertension, respiratory muscle weakening). Pulmonary involvement may be asymptomatic.
Patients with established PM/DM are at risk for infection. They are frequently immunosuppressed, and both opportunistic and nonopportunistic infections develop. In one study a majority of PM/DM patients (89%) exhibited an opportunistic infection at some time after the onset of their PM/DM. A variety of organisms were responsible for these infections, but more than half were due to fungi.
Pulmonary hypertension can develop in patients with PM/DM, and in concert with the myositis, which affects the respiratory muscles, the result may be markedly abnormal physiologic parameters even with relatively normal imaging results. The pulmonary hypertension seen in those with PM/DM is usually secondary to ILD, although primary pulmonary vascular disease has been described.
Weakness of the respiratory muscles may lead to respiratory failure; nevertheless, mild respiratory failure has a good prognosis and mechanical ventilation is rarely required for respiratory muscle weakness alone.
Weakness of the laryngeal musculature may cause dysphonia, and esophageal involvement is usually manifested as dysphagia. Esophageal disease is estimated to be present in up to half of patients with inflammatory arthropathy, and the esophageal myopathy can be proximal as a result of striated muscle involvement or distal as a result of smooth muscle involvement, particularly in overlap connective tissue disease.
Fulminant myositis can precipitate acute renal failure as a consequence of severe rhabdomyolysis and myoglobinuria.
Cutaneous eruptions, which occur in DM, tend to be dusky and erythematous. Periorbital edema with a heliotrope (purplish appearance) hue is pathognomonic for DM. The skin lesions frequently fade completely but may be followed by brownish pigmentation, atrophy, scarring, or vitiligo. Subcutaneous calcification may occur, particularly in childhood DM. This is similar in distribution to that encountered in systemic sclerosis but tends to be more extensive (calcinosis universalis), particularly in untreated or undertreated disease. Polyarthralgia, accompanied at times by joint effusions, joint swelling, and other evidence of nondeforming arthritis, occurs in about 30% of patients but tends to be mild.
Although symptomatic cardiac involvement is not common in PM/DM, it is of prognostic importance. Cardiac involvement is recognized as the third leading cause of death in this condition, after sepsis and malignancy. Various cardiac manifestations have been described in the literature, including conduction abnormalities, myocarditis, cardiomyopathy, coronary artery atherosclerosis, valvular disease, and pericardial abnormalities.
There is an association with malignancy and the inflammatory myopathies, but the association is strongest with adult-onset DM. Even though the link was refuted in an analysis of 102 patients with adult-onset DM, more recent evidence from population-based cohort studies has confirmed an association between malignancy and DM and PM. These studies show an even stronger association between PM and malignancy, with recent data confirming that the association of DM and PM with malignancy is not purely due to various biases. The majority of these malignancies are adenocarcinomas, with the strongest correlation with ovarian carcinoma. Other associations include cervical, lung, pancreas, bladder, and gastric adenocarcinomas. Of interest, the peak incidence of cancer diagnosis occurs within the first year of diagnosis of the myopathy. In some patients the diagnosis of cancer occurs during relapse of previously dormant myositis.
The classification of PM/DM is potentially confusing. Amyopathic DM is DM without muscle weakness, and although pulmonary fibrosis is uncommon in these patients, isolated case reports do appear. Antisynthetase syndrome occurs in a subgroup of inflammatory myopathy patients who have antisynthetase antibodies; anti–Jo-1 antibody is one of these antisynthetase antibodies. These patients have features similar to those of PM and DM, but they are more likely to have significant ILD. Antisynthetase syndrome is also associated with “mechanic’s hands,” a condition in which the skin of the patient’s fingers is thickened and cracked with accompanying polyarthritis and Raynaud phenomenon.
Pathophysiology
Pathology
The most common parenchymal lung diseases associated with PM/DM are nonspecific interstitial pneumonia (NSIP), organizing pneumonia (OP), diffuse alveolar damage (DAD), usual interstitial pneumonia (UIP), and alveolar hemorrhage (secondary to capillaritis). In a review of surgical lung biopsy specimens from patients with PM/DM, the predominant histologic process was NSIP. In another series by Douglas and coworkers, 70 patients with PM/DM and ILD were examined. In this group 22 patients underwent surgical lung biopsy, and the majority in this series had NSIP histologically (81%). The remaining patients had DAD, OP, and UIP.
PM/DM patients can progress through the pathologic sequence of DAD, then OP, and finally fibrotic NSIP. The first step is acute epithelial injury, and then organization occurs with the production of intraalveolar buds of granulation tissue (OP). On serial evaluation this patchy consolidation, which is presumed to be OP, may be partially reversible; alternatively, it may progress to reticular abnormality. OP and NSIP are frequently found together on lung biopsy specimens.
Acute ILD in patients with PM/DM may have a rapidly progressive course, and in a study of 81 patients with inflammatory myopathy, 5 had acute ILD and all 5 had died within 6 months of disease onset. These 5 patients had histologically proven DAD, and in all cases the muscle involvement was typical of DM but was classified as mild. Acute ILD in PM/DM has high mortality, and all the patients were seronegative for antisynthetase antibodies. Seronegativity for antisynthetase antibodies is considered a poor prognostic indicator in DAD associated with PM/DM.
Active and healed vasculitis has been demonstrated pathologically in PM/DM. Unlike rheumatoid arthritis and systemic lupus erythematosus, pleural disease is not commonly described, but fibrinous pleuritis has been recorded at autopsy (12%). Pleural effusions were also present in a quarter of cases and were presumed to be secondary to heart failure or malignancy.
Lung Function
Lung function testing is useful for the early diagnosis of pulmonary involvement in PM/DM. The earliest abnormality detected is usually a decrease in the diffusing capacity for carbon monoxide (DLCO). A restrictive pattern subsequently develops if pulmonary fibrosis occurs. An important parameter is K co , defined as the ratio of the rate of DLCO to the accessible alveolar volume (V a ). K co values less than 70% of predicted in a nonsmoker can be helpful in suggesting an occult vasculopathy. In patients with PM, half have a 50% reduction in respiratory muscle strength, and hypercapnia is likely to occur when respiratory muscle strength is less than 30% of normal. The hallmark of respiratory muscle weakness is a reduction in maximum inspiratory and expiratory pressure.
Manifestations of the Disease
Primary thoracic manifestations of PM/DM include one or more of the following ILDs: NSIP, OP, UIP, and DAD. Vascular manifestations include vasculitis, capillaritis/hemorrhage, and pulmonary hypertension. Secondary manifestations include respiratory failure, atelectasis, pneumonia, aspiration, drug-related disorders, heart failure, and various neoplasms.
Radiography
Evidence of ILD, especially OP, may precede the frank myositis and skin lesions. In PM/DM chest radiographs commonly demonstrate bilateral irregular linear opacities (reticulation) at the lung bases and occasionally consolidation. In most series of PM/DM the proportion of patients with evidence of ILD on chest radiography is approximately 10%. In one of the largest studies of PM/DM-ILD, abnormalities on chest radiography included irregular linear opacities (95%), consolidation (25%), honeycombing (4%), and pleural effusions (4%). These findings most commonly reflect the presence of NSIP, OP, or DAD.
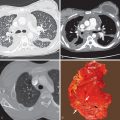
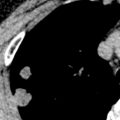
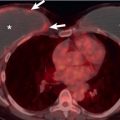
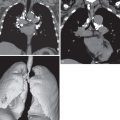
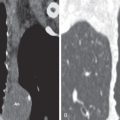
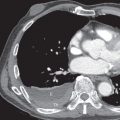
The most common ILD in patients with PM/DM is NSIP. The main radiographic manifestations of NSIP consist of a reticular pattern and hazy increased opacity (ground-glass opacities) in the lower lung zones ( Fig. 42.1 ). The most frequent manifestations of OP consist of bilateral areas of consolidation that may be patchy, mainly peribronchial or subpleural, and that may involve any lung zone ( Fig. 42.2 ). Patients with PM often have findings of both NSIP and OP radiologically and histologically ( Fig. 42.3 ). DAD is characterized by a sudden onset and rapid progression from ground-glass opacities to extensive bilateral consolidation. The main differential diagnosis of areas of consolidation is aspiration or pneumonia. Pleural disease is relatively uncommon in PM/DM. The majority of pleural effusions seen in these patients are related to heart failure, infection, or malignancy.