Name
Action
Abbreviation
Initial publication
[18F]Cyclofoxy
Antagonist, μOR, and κOR
–
Pert et al. (1984)
[11C]Diprenorphine
Antagonist, non-subtype selective
DPN
Luthra et al. (1985)
[18F]Diprenorphine
Antagonist, non-subtype selective
FDPN
Wester et al. (2000)
[11C]Buprenorphine
Mixed agonist/antagonist, non-subtype selective
BPN
Luthra et al. (1987)
[18F]-FE-PEO
Full agonist, non-subtype selective
–
Riss et al. (2013)
[11C]Carfentanil
Agonist, μOR
CFN
Dannals et al. (1985)
[11C]Methylnaltrindole
Antagonist, δOR
MeNTI
Lever et al. (1992)
[11C]GR103545
Agonist, κOR
–
Talbot et al. (2005)
[11C]MeJDTic
Antagonist, κOR
–
Poisnel et al. (2008)
[11C]LY2795050
Antagonist, κOR
–
Zheng et al. (2013)
[11C]NOP-1A
Antagonist, ORL1
–
Pike et al. (2011)
[18F]MK-0911
Antagonist, ORL1
–
Hostetler et al. (2013)
The first attempt at in vivo labeling of opioid receptors was made in 1975. Investigators at Johns Hopkins University administered a labeled opioid receptor antagonist, [3H]naloxone, to living rats. In the excised brain tissue of these animals, they observed a regional distribution of radioactivity which corresponded to the known distribution of opioid receptors in the rodent brain (striatum > hindbrain > cerebellum). Because of strong nonspecific binding of [3H]naloxone, acceptable target-to-nontarget ratios of radioactivity could only be reached after ex vivo washing of brain slices to remove nonspecifically bound tracer. Yet, this seminal study paved the way for all later imaging of neuroreceptors in the living brain (Pert and Snyder 1975).
The first successful PET probe for opioid receptors was prepared in 1984 (Channing et al. 1985; Pert et al. 1984). After intravenous administration of [18F]3-acetylcyclofoxy (3-acetyl-6-deoxy-6-ß-[18F]-fluoronaltrexone), a regional distribution of radioactivity was observed which corresponded to the regional distribution of opioid receptors in the rat and baboon brain. Bound tracer in the baboon brain was completely displaced by the active (−)enantiomer of naloxone, whereas an identical dose of the inactive (+)enantiomer (0.13 mg/kg) had no effect (Pert et al. 1984).
Since [3H]naloxone showed a low ratio of specific-to-nonspecific binding, an antagonist with a higher affinity ([3H]diprenorphine, [3H]DPN) was prepared and successfully applied in autoradiography (review in Frost (2001)). However, labeling DPN with 11C proved difficult. Because of these difficulties, radiochemists focused their attention on other opioid ligands, and a method was developed to label carfentanil (CFN) with 11C, using a carboxylic acid analog of carfentanil as a precursor (Dannals et al. 1985). The resulting tracer, [11C]CFN, was used to acquire the very first images of opioid receptors in the human brain (Frost et al. 1985). At baseline, a regional distribution of radioactivity was observed which corresponded to the known regional densities of μOR. After treatment of the volunteer with naloxone (1 mg/kg), cerebral uptake of the tracer was strongly suppressed, indicating that [11C]CFN binds to μOR in the living brain in vivo (Frost et al. 1985). A regional distribution of radioactivity consistent with μOR binding and blockade of tracer uptake by naloxone were also observed in the mouse brain. Thus, [11C]CFN is suitable for studies of μOR in rodents (Saji et al. 1992).
In the same year that the first images of opioid receptors in the human brain were acquired, radiochemists developed a method for preparation of [11C]DPN. The compound was labeled at the N-carbon atom of its cyclopropylmethyl group (Luthra et al. 1985). A study with [11C]DPN in four healthy volunteers indicated that the tracer is rapidly washed out from brain regions with a low density of opioid receptors, whereas activity is retained in receptor-rich target areas. Eighty to ninety percent of tracer binding in target regions is naloxone reversible, suggesting that [11C]DPN is suitable for studies of opioid receptors in man (Jones et al. 1988).
A different method to prepare 11C-DPN was later published. That method involves [11C]-O-methylation of 3-O–t-butyldimethylsilyl-(6-O-desmethyl)diprenorphine with [11C]methyl iodide, followed by acidic deprotection of the reaction product (Lever et al. 1987). The advantage of this approach compared to the previously published method is its greater radiochemical yield.
The non-subtype-selective opioid ligand buprenorphine (BPN) has also been labeled with 11C. The first published method involved the reaction of an N-(decyclopropylmethyl) precursor of buprenorphine with [11C]cyclopropanecarbonyl chloride followed by reduction with lithium aluminum hydride (Luthra et al. 1987). Later, a more facile synthesis of [11C]BPN was reported, in which the drug was labeled with 11C at the 6-methoxy position, using a methylation reaction with [11C]iodomethane (Lever et al. 1990). In a preclinical study in a baboon, the cerebral kinetics of [11C]BPN and [11C]DPN were directly compared. (−)Naloxone-sensitive binding of both ligands was observed in the striatum but not in the cerebellum. Uptake values and time courses in the target region were similar. The investigators concluded that [11C]DPN shows better kinetics than [11C]BPN since [11C]DPN is cleared more rapidly from brain areas devoid of opioid receptors (Shiue et al. 1991). [11C]BPN shows a regional distribution in the baboon brain consistent with regional opioid receptor density, whereas bound radioactivity in target areas can be displaced by naloxone. Moreover, [11C]BPN binding in the striatum shows a good (~10 %) short-term test-retest variability (Galynker et al. 1996).
A non-subtype-selective agonist radioligand for opioid receptor imaging has recently become available. This compound, called 18F-FE-PEO, binds with K i values of 0.4–1.6 nM to all opioid receptor subtypes. The distribution of radioactivity in the rat brain corresponds to regional opioid receptor levels, but nonspecific binding is rather high (≥33 % even in target areas). 18F-FE-PEO is relatively slowly metabolized (60 % parent in rat plasma after 3 h), and radioactive metabolites do not enter the brain (Riss et al. 2013).
The first successful radioligand for δOR was the antagonist N1′-([11C]methyl)naltrindole ([11C]MeNTI). [11C]MeNTI was synthesized by alkylation of 3-O-benzylnaltrindole with [11C]methyl iodide followed by removal of the protecting benzyl moiety. [11C]MeNTI binding in the brain of living mice was blocked (up to 75 %) by naltrindole (δOR antagonist), but not by cyprodine (μOR antagonist) nor by U50,488 (κOR agonist). Regional tracer uptake showed a linear, close correlation to δOR densities known from autoradiography. These data indicated that [11C]MeNTI binds selectively to δOR in the mouse brain in vivo (Lever et al. 1992).
An attempt to label the delta-1 subtype of δOR in the mouse brain with the selective ligand [3H]benzylidenenaltrexone (BNTX) was only partially successful. Although some specific binding of the compound was observed in vivo and tracer uptake could be blocked with the appropriate compounds, ratios of specific-to-nonspecific binding were very poor (Lever et al. 1996). Fluorinated analogs of naltrindole (N1′-fluoroethyl-naltrindole or BU97001 and N1′-fluoroethyl-(14-formylamino)-naltrindole or BU97018) have been proposed for PET imaging. BU97001 has been tritiated and used successfully for autoradiography of the rat brain; it shows sub-nM affinity to δOR (K d 0.42 nM) (Tyacke et al. 2002). However, PET data for [18F]BU97001 have not yet been reported.
A 11C-labeled N-substituted quinolinimide derivative with much higher subtype selectivity than [11C]MeNTI has been tested as a potential ligand for in vivo imaging of δOR. The compound was rapidly degraded in both plasma and brain tissue after intravenous injection in rats, and specific binding could not be detected in vivo (Bourdier et al. 2007). A radiofluorinated δOR antagonist based on the Dmt-Tic pharmacophore has also been prepared. Although the compound showed significant binding to δOR in brain slices, it failed to cross the blood–brain barrier in living rats (Ryu et al. 2008). Three benzamide δOR agonists have been labeled with 11C and subjected to preclinical testing. All compounds showed a very low brain uptake and no regional specific binding in mice. One of the compounds, called [11C]SNC80, was also tested in a monkey. Results in this primate were similar to those seen previously in mice, i.e., low blood–brain barrier permeability and a uniform distribution of radioactivity throughout the brain, indicating nonspecific binding (Pichika et al. 2010).
Initial attempts at developing subtype-selective radioligands for κOR were not successful. 18F-labeled analogs of U-50488 were prepared by fluoroalkyl substitution, but the labeling procedure resulted in a very strong (≥100-fold) loss of affinity with reference to the parent compound (Chesis et al. 1990b). A successful agonist radioligand for κOR, [11C]GR89696, was finally prepared in 1999 (Ravert et al. 1999). The R enantiomer, later called [11C]GR103545, shows an excellent hypothalamus/cerebellum ratio of 11.4 in the mouse brain at 90 min after injection, in contrast to the S enantiomer which is inactive (target/nontarget ratio = 1) (Ravert et al. 2002). Using subtype-selective opioid receptor ligands in blocking studies, the binding of [11C]GR103545 was shown to be κOR selective and saturable. Preclinical evaluation in baboons also indicated that [11C]GR103545 is a promising tracer for imaging of κOR. A regional distribution of radioactivity is observed that is consistent with the regional density of κOR. Naloxone pretreatment reduces tracer uptake in all brain regions to the level observed in the cerebellum. In vivo binding of the tracer is stereoselective, and significant washout occurs within the time frame of a PET scan (Talbot et al. 2005). A one-pot radiosynthesis for preparation of [11C]GR103545 with high specific radioactivity has recently been described (Nabulsi et al. 2011).
Antagonist radioligands for κOR have also been proposed. [11C]MeJDTic was prepared by methylation of the precursor JDTic with [11C]methyl triflate. In a preclinical study in mice, high uptake of the tracer was noted in target organs (lung, hypothalamus). Blocking and displacement studies with nonradioactive kappa, mu, and delta ligands suggested that [11C]MeJDTic binds specifically to κOR in the brain of living mice in vivo (Poisnel et al. 2008). Yet, no follow-up studies with [11C]MeJDTic have been reported. Another antagonist, [11C]LY2795050, was evaluated in Sprague–Dawley rats, wild-type and κOR knockout mice, and rhesus monkeys. Specific binding of the probe to cerebral κOR was observed; this binding was completely blocked by pretreatment of monkeys with naloxone (1 mg/kg) and dose-dependently inhibited by the selective κOR antagonist LY2456302. Tracer metabolism in primates is moderate (40 % parent remaining in monkey plasma at 30 min), and tracer pharmacokinetics appears favorable; thus, [11C]LY2795050 is the first successful antagonist radioligand for κOR (Zheng et al. 2013).
The last subclass of the opioid system for which imaging probes have been developed is the nociceptin/orphanin or opiate-like receptor (ORL1). [11C]Methyl-Ro64-6198 failed as a radioligand for this subtype since its in vivo binding in the mouse brain was largely nonspecific (Ogawa et al. 2001). A second probe, [11C]CPEB, showed some saturable in vivo binding but lack of specificity to ORL1 (Ogawa et al. 2003).
The first successful ORL1 ligand was [11C]NOP-1A, a specific antagonist with sub-nM affinity (0.15 nM). This compound was selected from a library of ORL1 antagonists developed by Eli Lilly & Co. (Pedregal et al. 2012). After intravenous administration of [11C]NOP-1A to rhesus monkeys, a regional cerebral distribution of radioactivity is observed consistent with binding of the probe to its target. Brain uptake is reduced by 50–70 % after pretreatment of animals with the ORL1 antagonist SB-612111 (Kimura et al. 2011; Pike et al. 2011). The regional distribution of [11C]NOP-1A in the human brain resembles that in the monkey brain. Regional distribution volume of the tracer can be reliably estimated using a 2-tissue compartment model with arterial sampling or Ichise’s noncompartmental bilinear analysis (Lohith et al. 2012).
Another ORL1 antagonist for PET imaging has recently been developed by Merck. Their compound [18F]MK-0911 shows a regional distribution in the monkey and human brain consistent with the regional expression of ORL1. Binding in the monkey brain is inhibited after pretreatment of animals with several structurally diverse ORL1 antagonists (MK-0584, MK-0337, MK-5757). Dose-dependent occupancy of ORL1 in the human brain by nonradioactive MK-5757 could be assessed with [18F]MK-0911 and PET (Hostetler et al. 2013).
20.3 Characteristics of Widely Used Radioligands
20.3.1 Cyclofoxy
[3H]Cyclofoxy, the parent compound of [18F]cyclofoxy, labels both μOR and κOR in the autoradiography of the rat and guinea pig brain (McLean et al. 1987; Rothman and McLean 1988). The regional pattern of radioactivity in the rat brain after in vivo injection of [3H]cyclofoxy corresponds to the regional distribution of these opioid receptor subtypes (Ostrowski et al. 1987; Rothman and McLean 1988), and brain radioactivity is reduced to background levels after pretreatment of animals with unlabeled naloxone (Ostrowski et al. 1987). K i values of (−)cyclofoxy for μOR, κOR, and δOR in rat brain membranes are 2.6, 9.3, and 89 nM, respectively, whereas the (+)enantiomer is essentially inactive (K i values > 10 μM) (Rothman et al. 1988). Consistent with these in vitro data, uptake of the radiofluorinated (−)enantiomer in the rat brain shows striking regional differences consistent with tracer binding to opioid receptors, whereas the tritiated (+)enantiomer is rather homogeneously distributed and shows low brain uptake. Pretreatment of animals with unlabeled (−)cyclofoxy reduces the uptake of [18F](−)cyclofoxy in all regions to the level of the tritiated (+) enantiomer (Kawai et al. 1990). Thus, [18F]cyclofoxy visualizes μOR and κOR in the living brain (Fig. 20.1).
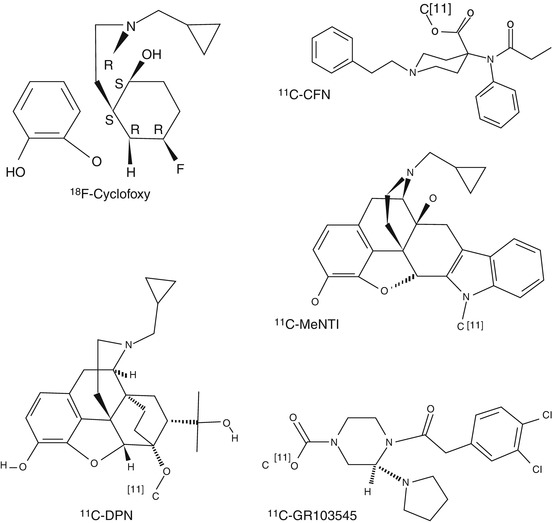
Fig. 20.1
Chemical structures of widely used opioid radioligands
Kinetic analysis of [3H]cyclofoxy uptake in the rat brain suggested an in vivo K d of 2.1–5.2 nM in different brain areas and a B max ranging from 1 pmol/g in the cerebellum to 78 pmol/g in the thalamus. Binding was reversible on a PET time scale (Sawada et al. 1991). In a follow-up study, a combination of bolus injection and constant infusion of [18F]cyclofoxy was applied to estimate the receptor-ligand parameters in a condition of “true” tissue-blood equilibrium, which was reached after 60 min. K d values of 1.4–2.9 nM were measured and B max in target areas ranged from 15 to 74 pmol/g tissue. Rodent cerebellum could not be used as a reference region since the level of nonspecific binding in the cerebellum was different from that in other parts of the brain (Kawai et al. 1991). Bolus plus continuous infusion of [18F]cyclofoxy was also advantageous in baboons, as it resulted in shorter scan times, a simplified protocol for blood sampling, and more accurate receptor measurements (Carson et al. 1993).
20.3.2 CFN
In vitro binding assays in human thalamic membranes indicated a K d of 0.08 nM for 3H-CFN (Titeler et al. 1989). In the rat brain, K i values of 0.051, 4.7, and 13 nM were measured at μOR, δOR, and κOR, respectively (Frost et al. 1985). For the guinea pig brain, the corresponding values were 0.024, 3.28, and 43.1 nM (Cometta-Morini et al. 1992). In vitro competition studies using opioid ligands with a wide range of subtype selectivities confirmed that [3H]CFN selectively labels μOR, in both the rat and human brain (Titeler et al. 1989). Thus, CFN is a potent and selective μOR agonist.
A study which applied compartment modeling for the analysis of 11C-CFN binding in the human brain showed that the occipital cortex can be considered as a region with low or negligible receptor density. Target region-to-occipital cortex ratios of radioactivity at late time points can be used instead of ratios of k 3/k 4 when tracer kinetic modeling is not feasible, e.g., in clinical research protocols (Frost et al. 1989). However, Logan graphical analysis or a simplified reference tissue model approach can provide more accurate estimates of 11C-CFN binding to μOR in the human brain than the ratio method (Endres et al. 2003). A test-retest study which assessed 11C-CFN binding in the brain of healthy volunteers twice, on a single day, indicated that both the regional two-tissue compartment model distribution volume (V T) of the tracer and binding potential (BPND) are reproducible (variability <6 and <10 %, respectively) (Hirvonen et al. 2009).
Administration of 11C-CFN can induce somnolence and sedation and, at higher dose, respiratory arrest. These pharmacological effects are related to the fact that CFN is an opioid agonist. The upper limit for the administered mass of CFN is about 0.1 μg/kg (Frost 2001). [11C]CFN binding in the human brain is affected by polymorphisms of the μOR gene. Carriers of the G allele (Asn40Asp variant, which may contribute to the development of alcohol dependence) have lower global binding potential than subjects homozygous for the A allele (Weerts et al. 2013).
Radiofluorinated analogs of carfentanil with subnanomolar affinities to μOR have been prepared (Henriksen et al. 2005b). N-[4-(methoxymethyl)-1-[2-(2-thienyl)ethyl]-4-piperidinyl]-N-phenyl-2-(±)-[18F]fluoropropan-amide ([18F]sufentanil) showed a high brain uptake in living mice and a distribution of radioactivity corresponding to regional μOR expression. The compound was rapidly metabolized in plasma (only 21 % parent remaining at 40 min), but the metabolites appeared to not enter the brain in significant amounts (Henriksen et al. 2005a).
20.3.3 DPN
Another widely applied radioligand for opioid receptor mapping is [11C]DPN. [3H]DPN binds with subnanomolar (K d 0.22–0.23 nM) affinity to opioid receptors in the rat brain (Chang et al. 1981) and in the human frontal cortex (Pfeiffer et al. 1982). K i values of DPN for μOR, δOR, and κOR in the rat brain are 0.20, 0.18, and 0.47 nM, respectively (Chang et al. 1981). Thus DPN is a non-subtype-selective opioid antagonist.
Using RB101, an inhibitor of enkephalin-degrading enzymes, the in vivo binding of [3H]DPN to opioid receptors in the mouse brain was shown to be sensitive to competition by endogenous enkephalins. Although RB101 is devoid of affinity to opioid receptors, the compound dose-dependently inhibits [3H]DPN binding in the unstressed mouse brain up to a maximum of 30 %. When mice were stressed by a warm-swim test, a greater inhibition of [3H]DPN binding by RB101 was observed (maximally 45 %). These effects of RB101 are probably related to increased levels of endogenous enkephalins (Ruiz-Gayo et al. 1992).
A validation study using [3H]DPN in rats showed that compartment modeling of PET data with the cerebellum as reference region can provide accurate measurements of regional opioid receptor B max and K d , although K d values estimated in vivo are an order of magnitude higher than those determined in vitro, probably due to diffusion gradients in tissue or protein binding of the radioligand (Cunningham et al. 1991). However, the cerebellum cannot be considered as a region devoid of opioid receptors in human studies since μOR and κOR are expressed at low levels in this area of the human brain (Schadrack et al. 1999).
The kinetics of [11C]DPN and [11C]CFN in the brain of human volunteers have been directly compared (Frost et al. 1990; Villemagne et al. 1994). Binding of both ligands in the thalamus (an area containing mainly μOR) was not significantly different, but [11C]DPN showed significantly greater binding than [11C]CFN in the striatum, cingulate, and frontal cortex. Dissociation of [11C]CFN from its binding sites was observed, but dissociation of [11C]DPN was hardly noticeable. [11C]CFN binding reached a plateau, whereas [11C]DPN uptake showed a steady increase (Frost et al. 1990). Total radioactivity in the human brain (SUV) and the ratio of total/nonspecific binding were twice as great after administration of [11C]DPN than [11C]CFN. The dose of naloxone required to fully block [11C]DPN binding was ten times greater than that required for [11C]CFN (Villemagne et al. 1994). Thus, [11C]DPN appeared to label δOR and ΚOR in addition to the μOR which are selectively labeled by [11C]CFN.
Both the regional density (B max) and radioligand affinity (K d) of opioid receptors can be assessed by applying a two-injection protocol, in which [11C]DPN is administered at high (20–160 TBq/mmol) and low (1–3 TBq/mmol) specific radioactivities (Sadzot et al. 1991) or by a pulse-chase protocol (Jones et al. 1994b). Parametric images of [11C]DPN binding can be prepared, using both conventional spectral analysis and rank shaping (Hammers et al. 2007b).
Radiofluorinated analogs of DPN have been prepared. N-(3-[18F]Fluoropropyl)-N-nordiprenorphine (FPND) showed rather limited brain uptake (Bai et al. 1990) but a striatum/cerebellum ratio of 3.3 ± 0.7 at 30 min postinjection which was reduced to unity after pretreatment of animals with naloxone (1 mg/kg). The tracer was extensively metabolized (<25 % parent remaining in blood at 30 min), but not significantly defluorinated (Chesis et al. 1990a). 6-O-(3-[18F]Fluoroethyl)-6-O-desmethyldiprenorphine ([18F]DPN) showed a regional binding pattern in the human brain similar to that of [11C]DPN (Wester et al. 2000). FDPN binds to μOR, δOR, and κOR in rat brain slices like the parent tracer, DPN (Wester et al. 2000). A scan duration of at least 90 min is required for reliable estimates of tracer distribution volume (Boecker et al. 2005). If metabolite-corrected plasma data are used as input function, individualized metabolite correction should be performed since women metabolize FDPN significantly faster than men (Henriksen et al. 2006).
20.3.4 MeNTI
K i values of MeNTI at δOR, μOR, and κOR in guinea pig brain membranes are 0.02, 14, and 65 nM, respectively (Portoghese et al. 1990). Thus, MeNTI is a very potent and subtype-selective δOR antagonist.
In the human brain, [11C]MeNTI shows regional kinetics concordant with selective binding of the tracer to δOR. Prolonged retention of radioactivity is observed in receptor-rich areas, whereas the ligand is rapidly washed out from nontarget areas of the brain. The regional distribution of radioactivity is closely correlated with local δOR densities but not with regional densities of μ or κ binding sites. Tracer uptake is blocked after pretreatment of volunteers with naltrexone (up to 73 %) (Madar et al. 1996).
[11C]MeNTI shows irreversible binding characteristics in the human brain during a 90-min scanning period. A constrained three-compartment kinetic model is superior to other data-analytical techniques for quantification of the in vivo binding of the tracer to δOR (Smith et al. 1999).
20.3.5 GR103545
The subtype selectivity of [11C]GR103545 has been determined in HEK-239 cells expressing human κOR, CHO-K1 cells expressing human μOR, and CHO cells expressing human δOR. K i values at the three OR subtypes were 0.02, 16.2, and 536 nM, respectively (Schoultz et al. 2010). Thus, [11C]GR103545 is an extremely potent and subtype-selective κOR agonist.
In a preclinical study in awake rhesus macaques, [11C]GR103545 showed a regional volume of distribution in the brain which corresponded to regional κOR densities (Schoultz et al. 2010). In a more recent study, the use of a bolus-plus-continuous-infusion protocol was shown to permit estimation of both B max and K d of κOR in the monkey brain in vivo (Tomasi et al. 2013). The cerebellum was used as the reference region in kinetic analysis.
A low injected mass of [11C]GR103545 is essential since this κOR agonist can induce respiratory depression, reductions of heart rate, and sedation even at very low administered doses of 0.3 μg/kg. Based on the K d value estimated for the radioligand in nonhuman primates, a dose of 1.4 μg (3.38 nmol) was estimated as an acceptable dose limit in human studies for subjects with a body mass of 70 kg (Tomasi et al. 2013).
20.4 PET Studies in Healthy Volunteers
20.4.1 Influence of Gender, Hormonal Status, and Age
11C-CFN-PET has been applied for the study of changes of opioid receptor binding in the brain of women during the menstrual cycle. No significant differences in tracer binding were noted between the follicular and luteal phases of the cycle, but follicular levels of estradiol in the circulation were negatively correlated with 11C-CFN binding potential in the amygdala and hypothalamus. These results, combined with measurements of luteinizing hormone pulse, suggested that μOR in the amygdala modulate gonadal steroid hormone pulsatility, whereas circulating estradiol regulates μOR function in the brain (Smith et al. 1998). Neuroimaging has shown that black cohosh, a herbal extract used to treat menopausal symptoms, affects the endogenous opioid receptor system. Significant increases in 11C-CFN binding potential (10–61 %) were noted in the cortical areas, thalamus, and nucleus accumbens of the brain of postmenopausal women (Reame et al. 2008). In the thalamus and nucleus accumbens, similar effects were observed after estrogen replacement therapy (Smith et al. 2006).
A PET study with 11C-CFN, involving 50 subjects, indicated that the cerebral binding of 11C-CFN is affected both by gender and by age. Binding potential of the radioligand increased with age in the neocortex and putamen. Women showed higher 11C-CFN binding than men of the same age in several cortical and subcortical areas; however, in postmenopausal women μOR binding in the thalamus and amygdala declined to levels lower than in age-matched men (Zubieta et al. 1999). A PET study with [18F]cyclofoxy confirmed that opioid receptor binding in the thalamus of females is lower than in males, both in healthy aged subjects and in patients with Alzheimer disease (Cohen et al. 2000). MRI-based partial volume correction is required for the accurate assessment of receptor densities in small structures of the human brain. Applying such correction to 11C-CFN data for the left temporal cortex, binding of the radioligand appeared to increase with age (0.9 % per year), consistent with B max values from postmortem autoradiography. Without partial volume correction, the effect of aging could not be observed (Bencherif et al. 2004a).
20.4.2 Personality Traits
Several PET studies have explored relationships between opioid receptor availability in the human brain and personality traits. Using the tracer [18F]DPN, German investigators scanned 23 healthy male volunteers under resting conditions. Subsequently, the subjects were psychologically tested for novelty seeking, harm avoidance, reward dependence, and persistence. A significant correlation was detected between the score for reward dependence (a personality trait predisposing for addictive behavior) and tracer binding potential in the bilateral ventral striatum with nucleus accumbens (Schreckenberger et al. 2008). A later study from the USA involved scanning of 19 young healthy male volunteers with the μOR ligand [11C]CFN and PET. Subjects were scanned both at baseline and during receipt of a painful stress challenge (infusion of hypertonic saline solution in the left masseter muscle). High scores for impulsiveness and low deliberation scores were associated with significantly higher baseline μOR availability and greater stress-induced endogenous opioid system activation, measured as a reduction of [11C]CFN binding potential. These differences were noted in several brain areas (prefrontal and orbitofrontal cortices, anterior cingulate, thalamus, nucleus accumbens, and basolateral amygdala). Thus, μOR binding potential appears to predict an individual’s vulnerability to risky behaviors, such as the development of substance abuse (Love et al. 2009). An interesting study from Finland measured brain μOR availability with [11C]CFN under resting conditions in 22 healthy volunteers: 10 individuals scoring in the upper and 12 individuals scoring in the lowest quartile for harm avoidance. A high score for harm avoidance (particularly in the subscales shyness with strangers, fatigability, and asthenia) was associated with high μOR availability (or low endogenous μ-opioid drive) in the anterior cingulate cortex, ventromedial and dorsolateral prefrontal cortices, and anterior insular cortex. Thus, high μOR availability may be related to vulnerability for affective disorders (Tuominen et al. 2012).
20.4.3 Affective Responses
The opioid system appears to play an important role in the regulation of affective responses. An initial PET study examined [11C]CFN binding and blood flow (uptake of 15O-water) in the brain of 12 healthy male volunteers, which viewed either aversive images (facial mutilation, wounds, dead bodies) or neutral images (common objects, benign scenes, normal faces). Higher baseline μOR binding potential in the left temporal pole of the subjects was associated with lower blood flow in this region during presentation of the aversive emotional stimuli. This finding was interpreted as evidence for an inhibitory or anxiolytic role of the opioid system in limbic regions of the brain (Liberzon et al. 2002).
A second study examined healthy female volunteers, using 11C-CFN and PET. Sustained neutral and sadness states were elicited by the recall of an autobiographical event associated with these emotions. Sustained sadness caused an increase of μOR availability in the rostral anterior cingulate, ventral pallidum, amygdala, and inferior temporal cortex which was interpreted as reflecting a reduction of the release of endogenous opioids due to deactivation of opioid neurotransmission (Zubieta et al. 2003).
Long-distance runners have often reported to enter a state of euphoria while running, commonly referred to as “runner’s high.” In order to examine possible mechanisms underlying this phenomenon, ten athletes were scanned with [18F]DPN and PET, both at rest and after 2 h of endurance running (in random order). By SPM2 analysis, reductions of tracer binding were noted in several brain areas after exercise. Reported levels of euphoria were inversely correlated with [18F]DPN binding in the prefrontal/orbitofrontal cortices, anterior cingulate cortex, bilateral insula, parainsular cortex, and temporoparietal regions. These findings support the idea that running is associated with increased release of endorphins, resulting in positive mood changes (Boecker et al. 2008).
Whereas sustained sadness in healthy volunteers is associated with a reduction in the release of endogenous opioids, positive emotion appears to be associated with an increase of that release. An interesting study from London scanned 25 healthy male volunteers with [11C]DPN and PET, both during positive mood induction (sexually oriented video clip, listening to favorite music, reading positive statements, unexpected gift of 30 GBP) and during neutral mood (nature movie, listening to unknown classical music, reading neutral statements, no gift). Significant reductions of [11C]DPN binding were noted in the hippocampus during positive mood induction, and the magnitude of positive mood change was negatively correlated with DPN binding in the amygdala (Koepp et al. 2009).
20.4.4 Pain
Opioid receptor expression ([11C]DPN or [18F]DPN binding) is high in projections of the medial and part of the lateral pain system of the human brain (Baumgartner et al. 2006; Jones et al. 1991). The lateral network is believed to be associated with the sensory aspects and the medial network with the affective aspects of pain perception. Reductions in receptor availability have been observed in healthy volunteers suffering pain. These data can be interpreted as decreases of regional receptor availability because of release of endogenous opioids, although internalization or downregulation of opioid receptors can also occur. Released opioids may mediate antinociception.
Sustained masseter muscle pain in 20 healthy volunteers was associated with significant declines of the binding of [11C]CFN in several cortical and subcortical brain regions (Zubieta et al. 2001). When capsaicin was applied to the dorsal side of the left hand of eight healthy volunteers, a pain-related decrease in [11C]CFN binding was measured in the contralateral thalamus (Bencherif et al. 2002). In eight healthy volunteers who received painful heat stimulation through the right forearm, a significant reduction of [18F]DPN binding was noted in the limbic and paralimbic areas of the brain, including the rostral anterior cingulate cortex and the insula (Sprenger et al. 2006a).
The duration of pain-induced changes in μOR binding potential has been examined in a PET study with [11C]CFN. Three subsequent 90-min scans were made in 14 healthy male volunteers; the first scan consisted of two 45-min baseline intervals, the second scan involved a 45-min period of sustained muscle pain followed by a 45-min baseline interval, and the third scan consisted again of two 45-min baseline intervals. Robust decreases of [11C]CFN binding were noted in several cortical and subcortical regions during the pain challenge. However, these changes did not persist in a subsequent scan. Pain-induced alterations of opioid release and/or μOR internalization are therefore short lasting (Scott et al. 2007b).
In a study involving 14 male and 14 female healthy individuals subjected to sustained masseter muscle pain (infusion of hypertonic saline), men demonstrated larger declines of [11C]CFN binding in the anterior thalamus, ventral basal ganglia, and amygdala. Conversely, women showed greater declines of the PET signal in the nucleus accumbens. Thus, opioid receptor-mediated antinociceptive responses show gender differences (Zubieta et al. 2002).
Opioid release is also involved in placebo analgesia. In a PET study with [11C]CFN, healthy subjects were scanned under four different conditions: painful skin heat with placebo cream, painful skin heat under control conditions, nonpainful warmth with placebo cream, and nonpainful warmth under control conditions. In all cases, the volunteers were told that the placebo cream was a powerful analgesic. Placebo treatment resulted in significant decreases of [11C]CFN binding in several brain regions known to be involved in pain and affect; thus, endogenous opioid release appears to be part of the mechanism by which expectancies regulate the perception of pain (Wager et al. 2007). A later study from the same group examined placebo and nocebo effects, i.e., the subjects expected the placebo cream to either ameliorate or exacerbate their pain. Placebo and nocebo effects were found to be associated with opposite opioid and dopaminergic responses. A placebo-activated opioid neurotransmission in the anterior cingulate, orbitofrontal and insular cortices, nucleus accumbens, amygdala, and periaqueductal gray, whereas dopaminergic neurotransmission was activated in the ventral basal ganglia including the nucleus accumbens. Nocebo responses consisted of a deactivation of opioid and dopamine release. The magnitude of opioid and dopamine activation was correlated with the anticipated and subjectively perceived placebo effect (Scott et al. 2008).
When heat pain was combined with either acupuncture or placebo needle stimulation in healthy volunteers, acupuncture was shown to result in altered binding of [11C]DPN (mainly decreases) in the orbitofrontal cortex, medial prefrontal cortex, insula, thalamus, and anterior cingulate cortex (Dougherty et al. 2008).
Opioid receptor availability at baseline conditions appears to be a predictor of human sensitivity to both painful (cold pressor test) and mechanical stimuli (touch, vibration). Greater sensitivity was associated with a lower BP of [18F]DPN in various cortical areas (Mueller et al. 2010). In a later study on 12 healthy male volunteers, a significant correlation was observed between baseline binding potential of [11C]CFN (μOR binding) in the striatum and the cold pressor pain threshold (Hagelberg et al. 2012).
20.4.5 Vestibular Processing
An interesting study examined involvement of the opioid system in vestibular neurotransmission in humans. Ten right-handed healthy volunteers were scanned with the ligand [18F]DPN, both under baseline conditions and during caloric stimulation of the right ear. Each 90-min scan session consisted of three subsequent phases: a prestimulation period (30 min), a stimulation or sham period (30 min), and a follow-up period (30 min). During the stimulation period, warm (44 °C) and cold (30 °C) air were alternated at 5-min intervals in order to avoid habituation; during the sham period the tubing was fixed to the subjects head in such a way that the air flow passed beside the ear. A decrease in tracer binding was noted in the right posterior insular cortex and the postcentral region during stimulation, indicating release of endogenous opioids and involvement of the opioid system in vestibular processing (Baier et al. 2010).
20.4.6 Myocardial Opioid Receptors
Both 11C-N-methyl-naltrindole (11C-MeNTI) and 11C-CFN show specific binding in the human heart. However, the reduction of myocardial distribution volume and binding potential of these opioid ligands after pretreatment of volunteers with naloxone is only minor (14–21 and 19–25 %, respectively), suggesting low levels of δOR and μOR expression and poor signal-to-noise ratios in PET (Villemagne et al. 2002).
20.4.7 Occupancy Studies
PET offers the unique opportunity of measuring the fraction of receptor populations occupied by nonradioactive drugs in the living brain and relating measured levels of occupancy to the magnitude of the therapeutic effect or to unwanted side effects. Receptor occupancy is estimated by assessing competition of a nonradioactive drug with the radioligand for the same binding sites. The basic idea was already published in 1980 (Homcy et al. 1980).
Using the radioligand [11C]CFN, a half-life of μOR blockade of 72–108 h was measured in the brain of healthy volunteers after a single oral dosing of naltrexone, corresponding to the duration of pharmacological effects when the drug is used for treatment of heroin dependence (Lee et al. 1988).
A later study with 11C-CFN compared the duration of occupancy of μOR in the human brain by the antagonists nalmefene and naloxone. Clearance half-lives were 28.7 h after administration of 1 mg of nalmefene and 2.0 h for 2 mg of naloxone. The longer blockade by nalmefene could represent an advantage in the clinical use of this compound for reversal of opioid anesthesia or for treatment of drug addicts after an opioid overdose (Kim et al. 1997). Nalmefene has also been proposed for treatment of alcoholism. The decline of μOR occupancy in the human brain after single or repeated nalmefene dosing was found to be slower than the decline in plasma concentration of the drug or its metabolites, indicating slow dissociation of receptor-bound nalmefene in vivo (Ingman et al. 2005).
Buprenorphine is used for the treatment of heroin dependence. Buprenorphine occupancy of μOR in the human brain has been examined, using 11C-CFN and PET. Four hours after administration of 2 mg buprenorphine, receptor occupancy ranged from 36 to 50 %. When the buprenorphine dose was raised to 16 mg, levels of occupancy increased to 79–95 %. Placebo-treated heroin abusers had greater μOR binding potential in the inferofrontal and anterior cingulate cortex compared to matched healthy controls (Zubieta et al. 2000). Increasing doses of buprenorphine were associated both with decreased μOR availability and with decreased withdrawal symptoms. The highest dose of buprenorphine (32 mg) caused virtually complete μOR occupancy (Greenwald et al. 2003). The duration of action of buprenorphine was examined in ten heroin-dependent subjects after termination of a 16-mg/day dose of the drug. Relative to a control group of heroin-dependent subjects maintained on placebo, μOR occupancy was 70, 46, 33, and 18 % at 4, 28, 52, and 76 h after last use of the drug. Receptor occupancy was correlated with withdrawal symptoms; an occupancy of 50–60 % appeared necessary for adequate symptom suppression (Greenwald et al. 2007).
Although the dose- and time-dependent opioid receptor occupancy by antagonists and partial agonists can be assessed with PET, measurements of receptor occupancy by full agonists may be problematic. No difference in [11C]DPN binding was found in the brain of opioid-dependent subjects stable on methadone (18–90 mg daily, PET scan performed at peak plasma levels of the agonist) as compared to healthy controls. In rats receiving increased doses of methadone, no dose-dependent reduction of the cerebral binding of [11C]DPN was observed. These negative findings were explained by assuming that methadone is already efficacious at very low levels of receptor occupancy (<10 %, i.e., below the detection limit of PET) (Melichar et al. 2005). In a subsequent preclinical study, rats were pretreated with antinociceptive doses of oxycodone (μOR and κOR agonist), morphine (μOR agonist), and buprenorphine (δOR and κOR antagonist, μOR partial agonist). Full agonists did not reduce the brain uptake of [11C]DPN at all, whereas buprenorphine administration resulted in a strong reduction (up to 90 %) of tracer binding (Hume et al. 2007). Opioid receptor occupancy by the antagonist naloxone could also be measured with [11C]DPN and PET, a drug dose of 13 μg/kg resulting in 50 % occupancy (Melichar et al. 2003). The reason for the negative findings with exogenous full agonists is not clear, since endogenous enkephalins have been shown to compete with [11C]DPN for OR binding in the human brain (Ruiz-Gayo et al. 1992). Moreover, an early PET study reported 19–32 % lower binding of [18F]cyclofoxy in various brain regions of long-term methadone-treated former heroin addicts as compared to controls (22 h after the last dose), and the reduction of tracer binding in caudate and putamen correlated with levels of methadone in plasma (Kling et al. 2000). Since the observed competition is both tracer and agonist dependent, these PET data may indicate the presence of multiple, different agonist binding sites on opioid receptor proteins or upregulation of subclasses of opioid receptors in heroin-dependent subjects.
Opioid receptor (particularly μOR) agonists increase and antagonists decrease feeding and other rewarding behaviors in animal models. A [11C]CFN-PET study examined cerebral μOR occupancy in 26 healthy male volunteers after single oral doses of either GSK1521498 (μOR inverse agonist, candidate drug for the treatment of overeating) or naltrexone (μOR and κOR antagonist). Activation of the amygdala by a palatable food stimulus (fruit drink) was examined in the same volunteers, using fMRI. Although a dose-dependent occupancy of μOR was observed for both compounds, GSK1521498 significantly attenuated activation of the amygdala by the food stimulus, in contrast to naltrexone. This difference may be related to the fact that μOR and κOR signaling have different effects on feeding behavior and reward processing (Rabiner et al. 2011).
Naloxone administration triggers activity of the hypothalamic-pituitary-adrenal (HPA) axis by blocking opioid inhibitory tone at corticotropin-releasing factor (CRF) neurons in the paraventricular nucleus of the hypothalamus. These neurons regulate the secretion of adrenocorticotropic hormone (ACTH) and cortisol. An interesting study measured μOR binding potential in the brain of 18 healthy subjects using [11C]CFN and PET. The following day, ACTH and cortisol responses to an incremental naloxone challenge were tested in the same subject group. Cortisol responses to naloxone were negatively correlated to [11C]CFN binding potential in several brain areas (ventral striatum, putamen, caudate, hypothalamus). Apparently, subjects with higher binding potential have lower endogenous occupancy of μOR by ß-endorphin and thus place less inhibitory tone on the HPA axis; therefore these subjects show a smaller cortisol response to naloxone (Wand et al. 2011).
20.5 PET Studies in Patients and Drug Addicts
20.5.1 Major Depressive, Borderline Personality, and Posttraumatic Stress Disorder
Using the sustained neutral and sadness state paradigm, μOR availability in the brain of healthy female volunteers and patients with major depressive disorder (MDD) has been compared. Healthy controls showed an increase of μOR availability in the anterior cingulate during the sustained sadness condition indicating deactivation of μOR-mediated neurotransmission, but in patients who did not respond to antidepressant treatment after PET imaging, sustained sadness was associated with a decreased binding of 11C-CFN in this region, i.e., activation of the opioid system. Patients with MDD showed significantly lower μOR binding potential in the posterior thalamus than healthy controls during the neutral state. These PET data indicated that endogenous opioid neurotransmission is altered in MDD (Kennedy et al. 2006). A later study from the same group showed that in MDD patients but not in healthy subjects, plasma levels of the inflammatory cytokine IL-18 are positively correlated with baseline μOR binding potential and with activation of μOR-mediated neurotransmission during a sadness challenge (Prossin et al. 2011). Thus, there may be a link between peripheral stress-activated proinflammatory mechanisms and central stress-activated neurotransmitter systems, such as the opioid system.
A later study from the same group examined responses of the μOR system to sustained sadness in patients with borderline personality disorder. During the baseline (neutral) state, patients showed greater [11C]CFN binding than healthy controls in the bilateral orbitofrontal cortex, caudate, and nucleus accumbens besides the left amygdala, but lower binding in the posterior thalamus. Induction of sadness caused greater declines of [11C]CFN binding potential in the pregenual anterior cingulate, left orbitofrontal cortex, left ventricular pallidum, left amygdala, and left inferior prefrontal cortex of the patients than of healthy controls and greater increases of binding potential in the left nucleus accumbens, the hypothalamus, and the right hippocampus/parahippocampus. Apparently, the opioid system of patients with borderline personality disorder differs from healthy, age- and sex-matched control subjects both at baseline and in its response to a negative emotional challenge (Prossin et al. 2010).
An interesting study compared μOR binding in the brain of 14 healthy male volunteers, 15 males with combat exposure but without posttraumatic stress disorder (PTSD), and 16 male patients with PTSD, using [11C]CFN and PET. The two groups which had been exposed to war trauma showed lower μOR binding in the amygdala, nucleus accumbens, dorsal frontal cortex, and insular cortex than healthy volunteers but higher μOR binding in the orbitofrontal cortex. PTSD patients showed a significant reduction of μOR binding in the anterior cingulate cortex compared with both other groups. Combat-exposed subjects without PTSD had lower μOR binding in the amygdala but higher binding in the orbitofrontal cortex than either PTSD patients or healthy controls. Thus, the opioid system of the brain shows specific changes both after trauma and in PTSD (Liberzon et al. 2007).
20.5.2 Pain
In a group of four patients with rheumatoid arthritis, regional cerebral opioid receptor binding of [11C]DPN was quantified both during a period of pain and during a period when the subjects were out of pain. Significant decreases of [11C]DPN binding were seen in many brain areas when subjects suffered pain (particularly straight gyrus and frontal, cingulate, and temporal cortex) (Jones et al. 1994a). In a patient with central poststroke pain which developed after a small pontine hemorrhagic infarction, a reduction of the binding of [11C]DPN was also noted, and this reduction was more striking than the hypometabolism of [18F]FDG on the lateral cortical surface contralateral to the symptoms (Willoch et al. 1999).
Changes of [11C]DPN binding in the brain of six patients after surgical relief of trigeminal neuralgia pain were examined in a later study. The volume of distribution of the tracer in several cortical areas (prefrontal, insular, perigenual, midcingulate, inferior parietal), basal ganglia, and the thalamus was found to be significantly increased after surgery (Jones et al. 1999). These data and the findings in the previous studies were interpreted as evidence for the release of opioid peptides during various forms of pain resulting in a reduction of the fraction of receptors available for ligand binding. However, changes of opioid receptor numbers (upregulation after surgery or after the disappearance of pain) could not be ruled out.
In a later study involving four patients with central neuropathic pain (mainly poststroke) and age-matched controls, patients were found to have significantly less [11C]DPN binding in the dorsolateral, anterior cingulate, and insula cortices, the thalamus, and the inferior parietal cortex. These reductions were not a direct consequence of their cerebral lesions, since these were located in other brain areas. The authors interpreted the PET data as evidence for reduced damping of nociceptor activity in the patients and as a possible explanation for the fact that high doses of opiates are frequently required to achieve optimal analgesia in subjects with central neuropathic pain (Jones et al. 2004). Essentially similar findings were reported in a study from another institution, using the same tracer and involving five patients with central poststroke pain and 12 healthy volunteers (Willoch et al. 2004).
An interesting study compared changes of [11C]DPN binding in the brain of patients with peripheral neuropathic pain and central poststroke pain. Peripheral neuropathic pain patients showed bilateral and symmetric decreases of [11C]DPN binding, in contrast to central poststroke pain patients where an asymmetric decrease was noted, which was most prominent in the contralateral hemisphere. The symmetric and bilateral decline in the former patient group may reflect endogenous opioid release, whereas the lateralized decrease in the latter group suggests a loss of opioid receptors (Maarrawi et al. 2007a). Thus, central and peripheral forms of neuropathic pain may have different effects on the opioid system of the brain.
In a group of ten patients with restless legs syndrome (RLS), significant negative correlations were observed between [11C]DPN binding in areas serving the medial pain system and RLS severity or pain scores. These findings suggested release of endogenous opioids within the medial pain system related to the severity of the syndrome (von Spiczak et al. 2005).
Changes of the opioid system in patients with cluster headache were also examined, using the tracer [11C]DPN and PET. When patients as a group were compared with healthy volunteers, a striking (>50 %) decrease of tracer binding was noted in the pineal gland but not in any other brain area. Within the patient group, opioid receptor availability in the ipsilateral hypothalamus and anterior cingulate cortex was negatively correlated to the duration of the headache disorder. The patients did not experience acute pain during or immediately before the study; thus, the observed alterations of [11C]DPN binding appear not to represent an effect of acute pain processing but rather alterations in the opioid system related to the development of the disease. Opioidergic dysfunction in circuitries generating the biologic clock may be a mechanism underlying cluster headache (Sprenger et al. 2006b).
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 Progress in PET Imaging of the Norepinephrine Transporter System
Progress in PET Imaging of the Norepinephrine Transporter System
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 PET and SPECT Imaging of Steroid Hormone Receptors
PET and SPECT Imaging of Steroid Hormone Receptors
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


