Fig. 17.1
Representative autoradiograms of rat (upper row) and neonate pig brains (lower row) obtained with an α7 nAChR ligand ([125I]α-bungarotoxin) and the semi-selective α4β2 ligand [3H]epibatidine. For anatomic reference, the right-hand side of the figure shows comparable sagittal planes modified from the rat brain atlas (Paxinos and Watson 1998) and the pig brain atlas (Felix et al. 1999). Structures marked in the reference atlases are Co cerebral cortex, Cd caudate nucleus, Cs superior colliculus, Cb cerebellum, Hi hippocampus, Th thalamus, Po pons, Me medulla oblongata
The aspects of receptor imaging in vivo as discussed in this review relate exclusively to the use of radiolabelled receptor ligands, although we note that optical imaging has been used to investigate the cholinergic system (Prakash and Frostig 2005). Furthermore, clinical imaging of α4β2 nAChR in human brain is reviewed elsewhere in this volume (see Sabri et al. in: PET and SPECT in Neurology). Therefore, the current review is focussed primarily on preclinical aspects including parts on instrumentation and radiotracer development.
17.2 Advances in Animal PET and SPECT Technology
The abundance of nAChRs can be measured in vitro and ex vivo using selective radioligands in conjunction with quantitative autoradiography. However, molecular imaging with positron emission tomography (PET) or single photon emission computed tomo-graphy (SPECT) makes possible the detection of neuroreceptors in the living brain. Contrary to autoradiography in vitro, in vivo imaging procedures allow for longitudinal studies in individual animals, thereby minimising issues arising from intersubject variability and allowing intervention or challenge studies. Until recently, animal PET studies were carried out using clinical scanners, most of which having a spatial resolution of approximately 5 mm; this is hardly adequate for resolution of structures within rodent brain. Only in the past 10 years have dedicated small-animal PET scanners become widely available. The best current instruments can attain a spatial resolution of little more than 1 mm full width at half maximum (FWHM) in the centre of the field of view, which is adequate to resolve structures as small as the mouse striatum.
PET technology is based upon coincident detection of two gamma photons (511 keV) arising from positron-electron annihilation subsequent to positron emission by decay of a neutron-deficient radionuclide incorporated in the tracer molecule. As such, the positron range prior to annihilation ultimately limits the spatial resolution of small-animal PET. The positron range is somewhat greater for positrons arising from the decay of carbon-11 than for the less energetic positrons emitted by fluorine-18. Iterative image reconstruction algorithms can approach a final resolution for 18F sources of little more than 0.7 mm FWHM (Reader 2008).
Some of the dedicated small-animal scanners such as the Focus 120 (Siemens) are designed to be readily movable between research units (Tai et al. 2001). Other systems, e.g. the microPET P4, Focus 220 (Siemens) and ClearPET (Raytest), can accommodate rodents and small non-human primates, whereas the larger Hamamatsu SHR-7700 instrument is designed specifically for non-human primate studies. The aperture and field of view of PET imaging systems designed for animal studies allow simultaneous recordings from up to eight mice (Rominger et al. 2010a), which should present a considerable economy in the experimental design of studies with transgenic animals. The most recent generation of preclinical PET scanners, such as the Inveon® PET-CT (Siemens), affords a considerable advantage in terms of anatomic information provided by the CT modality, but at the expense of lesser mobility for the hybrid scanners.
Small-animal PET and SPECT images are frequently characterised by a lack of anatomic information. In order to interpret the emission images, it has generally been necessary to employ some manual procedure for registration of the emission images to digitised brain atlases, based on histology or magnetic resonance imaging (MRI) atlases for rodent brain (Jupp et al. 2007; Rominger et al. 2010b). Contemporary multimodal imaging systems combine small-animal PET with SPECT, X-ray computed tomography (CT) and/or MRI. As noted above, PET-CT presents a great advantage for brain studies in that a high-resolution structural brain image in perfect registration with the PET image is obtained for each individual animal, rather than resorting to some standard atlas. In addition, the CT scan can be used to correct the PET images for attenuation by tissue, thus providing absolute quantitation of radioactivity concentrations in the brain without requiring an additional time-consuming transmission scan. MRI offers better tissue contrast than CT, but the combining of PET and MRI instrumentation has presented a greater technical challenge, due to incompatibility of traditional electronic components with a strong magnetic field. Nonetheless, a number of prototype PET/MRI scanners have been constructed, including the split magnet design in an animal microPET(R)MR system at the University of Cambridge (Lucas et al. 2007). A commercial PET/MRI system for rats and mice was developed by Mediso; its 1 T permanent magnet limits MRI applications but offers great flexibility for animal PET studies.
At the cutting edge of small-animal imaging is the head-mounted PET detector developed at Brookhaven (“ratCAP”), which is compatible with imaging in awake, behaving rats (Schulz et al. 2011). This technology promises to revolutionise preclinical molecular brain imaging, by avoiding the well-known pharmacological confounds presented by anaesthesia. The ratCAP has recently been tested within the bore of a very high-field microMRI scanner, with minimal loss of sensitivity arising from the requirement for gating of the PET acquisition (Maramraju et al. 2011). Likewise, a SPECT camera which can be placed within an MRI magnet has recently been developed for small-animal studies (Meier et al. 2011). Whereas attenuation correction is usually obtained by CT scanning, MR-based attenuation correction has been demonstrated for PET and SPECT imaging using clinical scanners (Marshall et al. 2011) but has yet to be developed for small-animal studies. Although this technology remains at an early stage of development, hybrid PET-SPECT should eventually be permissive to multiple tracer studies with simultaneous acquisitions.
17.3 PET and SPECT Radioligands Targeting nAChR
Many autoradiographic studies of nAChRs particularly in human brain have focussed on comparing the agonist-sensitive sites using rather non-selective ligands such as [3H]acetylcholine, [3H]nicotine, [3H/125I]epibatidine or [3H]cytisine with those of the α7-selective antagonist [3H/125I]α-bungarotoxin (Paterson and Nordberg 2000). This approach suggested the existence of at least three native receptor subtypes with multiple affinity states, such that the α-bungarotoxin-sensitive receptors predominantly represent homomeric α7 nAChRs, whereas the acetylcholine/nicotine binding sites represent mainly heteromeric α4β2 nAChRs and a small population of heteromeric receptors containing the α3 subunit. Given the predominance α4β2 and the α7 subtypes in the brain as documented above, they have presented the main targets for molecular imaging of nAChRs. The available information on the distribution, density and functional role of other subtypes in the brain is relatively sparse (Gotti et al. 2006; Sharma and Vijayaraghavan 2008). Furthermore, the current emphasis on α4β2 and the α7 subtypes as targets for pharmaceutical development may account for the paucity of selective high-affinity drugs for the other subtypes (Gündisch and Eibl 2011).
17.3.1 Radioligands for α4β2 nAChRs
Reviews summarising the history of ligand development for the nAChRs (Sihver et al. 2000a, b; Ding and Fowler 2005; Horti and Villemagne 2006; Horti et al. 2010) have helped galvanise research groups to generate new lead compounds for molecular imaging and therapeutics. Radiolabelled imaging agents successfully applicable for the quantitative visualisation of neuronal nicotinic receptors in living systems have to fulfil a multitude of criteria: (1) high target affinity in the subnanomolar range and specificity towards nontarget nAChR subtypes, (2) low non-specific binding and absence of confounding metabolism generating brain permeant radiometabolites, (3) rapid clearance from non-specific areas and plasma to reduce background in the target tissue and (4) high membrane permeability as well as (5) high permeability and low efflux at the blood–brain barrier.
The vast majority of novel tracers for α4β2 nAChRs have been derived from three compounds: nicotine, epibatidine and 3-piridyl ether. Four of these compounds have so far been used successfully to image α4β2 receptors in human brain (Sihver et al. 2000b): nicotine (Nordberg 1993); the two halogen-substituted derivatives of A-85380, namely, 2-fluoro-A-85380 (Kimes et al. 2003) and 6-fluoro-A-85380 (Horti et al. 2000); and the epibatidine derivative flubatine (Sabri et al. 2011). Receptor-ligand interactions frequently entail stereoselective features (Smith and Jakobsen 2007), as has been formally demonstrated for the case of nAChRs in human PET studies with the two stereoisomers of [11C]nicotine (Nordberg et al. 1991; Nordberg et al. 1992) and also several of the radioligands discussed below. A summary of the compounds that have been investigated for imaging of α4β2 nAChRs is given in Table 17.1.
Table 17.1
Results of biodistribution ex vivo and PET imaging studies in vivo of the cerebral binding of α4β2 nAChR-selective radioligands
Main findings | |
|---|---|
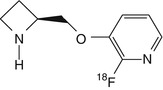 2-[18F]fluoro-A-85380 | Thalamic uptake of radioactivity in rat and baboon peaked at 60 min. In humans, cerebral uptake pattern consistent with the known distribution of α4β2 nAChRs in human brain. Total distribution volume significantly higher in smokers than in non-smokers, except the thalamus. Radioactivity in the brain reached steady state by 6 h (Dollé et al. 1999; Kimes et al. 2003, 2008; Mukhin et al. 2008) |
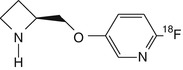 6-[18F]fluoro-A-85380 | In baboon dynamic PET, faster peak uptake and clearance as well as higher thalamus-to-cerebellum ratios than that obtained for 2-[18F]fluoro-A-85380 (Ding et al. 2000b) |
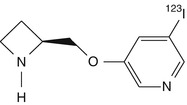 5-[123I]iodo-A-85380 | In rhesus dynamic PET, regional distribution in the brain consistent with the known nAChR distribution pattern. Relatively slow kinetics, with maximal binding ratios at more than 4 h. In baboon, significant displacement of radioactivity from cerebellum by cytosine, indicating this region inappropriate as reference region (Chefer et al. 1998; Fujita et al. 2000) |
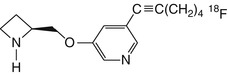 [18F]-ZW-104 | In baboon dynamic PET, rather slow kinetics in thalamus. High affinity towards multiple β2-containing nAChR subtypes (Valette et al. 2009) |
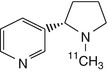 S-[11C]nicotine | In human and rhesus dynamic PET, binding was less selective for nAChR subtypes than was [11C]MPA (Sihver et al. 1999b) |
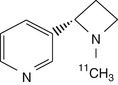 [11C]MPA | Specific binding in rhesus monkey proven by nicotine displacement in dynamic PET, but specific binding rather low (Sihver et al. 1999b) |
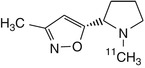 [11C]ABT418 | |
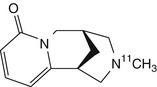 N-[11C]methylcytisine | No evidence of specific binding in rhesus monkey brain by dynamic PET (Valette et al. 1997) |
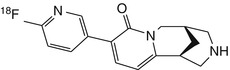 [18F]FPyCYT | In rat biodistribution study, low (0.3 % ID/g) and uniform brain uptake, with little evidence of specific binding (Roger et al. 2003) |
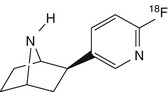 [18F]NFEP | |
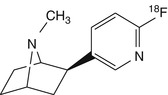 [18F]N-methyl-NFEP | In baboon dynamic PET, higher peak uptake in all brain regions than for [18F]NFEP. Despite milder toxicity than [18F]NFEP, evidence against safe use in humans (Ding et al. 1999) |
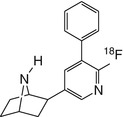 [18F]FphEP | |
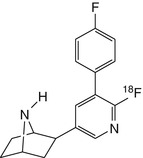 [18F]F2PhEP | Dynamic PET in baboon did not indicate reduction in brain distribution volume following pretreatment with nicotine (Valette et al. 2007) |
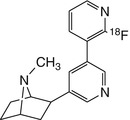 [18F]XTRA | Dynamic PET showed peak activity in baboon thalamus at 75 min after bolus, requiring several hours for steady-state measurement (Horti and Wong 2009) |
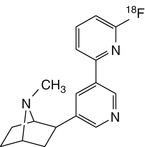 [18F]AZAN | |
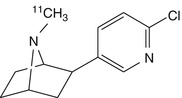 N-[11C]methylepibatidine | Different kinetics of brain uptake and washout seen for the stereoisomers in rats and mice. Dynamic PET showed high enrichment of radioactivity in the thalamus of the pig, but steady state not attained during 60 min scans. Applications in humans limited by high toxicity (Patt et al. 1999; Spang et al. 2000) |
 N-[11C]methylhomoepibatidine | In pig dynamic PET, the (−)-enantiomer showed a regional distribution and high accumulation in the thalamus consistent with representative for the α4bβ2 nAChR, which could be displaced by cytisine. Distribution of the (+)-enantiomer, non-specific. Applications in humans limited by high toxicity (Patt et al. 2001) |
 [11C]2-(6-chloro-5-phenylpyridin-3-yl)-methyl-7-azabicyclo[2.2.1]heptane | Rat biodistribution study showed high displaceable binding. Dynamic baboon PET showed rapid peak in thalamus, but increasing ratio relative to cerebellum over at least 2 h (Huang et al. 2004) |
 [11C]NMI-EPB | |
 [18F]flubatine | |
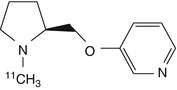 [11C]-A-84543 | Mouse biodistribution showed high brain uptake and a distribution consistent with the density of α4β2 nAChRs (Kassiou et al. 1998) |
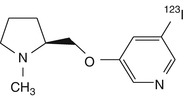 5-[123I]iodo-A-84543 | |
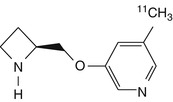 5-[11C]methyl-A-85380 | Mouse biodistribution consistent with α4β2 nAChRs. Rhesus monkey dynamic PET showed high non-specific binding (Iida et al. 2004) |
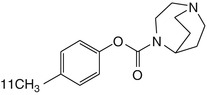
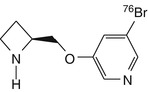 [76Br]BAP | In comparison to rat biodistribution, baboon dynamic PET showed higher non-specific binding with nicotine of cystine displacement (Sihver et al. 1999a) |
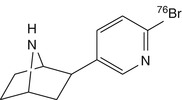 [76Br]BrPH | Rat biodistribution and baboon dynamic PET showed binding to nAChRs, but without subtype selectivity (Kassiou et al. 2002) |
 [11C]Me-p-PVC | Rapid accumulation in mice ex vivo, and rhesus PET studies, thus quantifiable with 2-h recordings, but BPND slightly lower than for 2-[18F]fluoro-A-85380 (Brown et al. 2004) |
 [11C]JHU85270 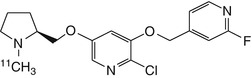 [11C]JHU85208 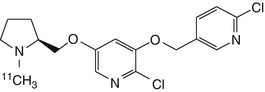 [11C]JHU85157 | In baboon dynamic PET, thalamic BPND lower than that of 2-[18F]fluoro-A-85380 (Gao et al. 2009) |
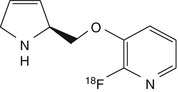 [18F]nifene | In rhesus dynamic PET, fast kinetics with peak thalamic binding at less than 10 min, thalamus-to-cerebellum ratio of ~2 (Pichika et al. 2006) |
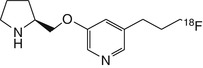 [18F]nifrolidine | In rhesus dynamic PET good labelling of thalamus, with slightly faster kinetics than for 2-[18F]fluoro-A-85380, with maximal binding at 70 min and with plateau thalamus-to-cerebellum ratio of 1.7 at 2 h (Chattopadhyay et al. 2005) |
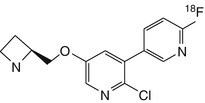 [18F]NIDA52189 | In rhesus dynamic PET, distribution consistent with α4β2 nAChRs, but BPND 2.5 times higher than for 2-[18F]fluoro-A-85380 (Zhang et al. 2004) |
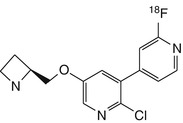 [18F]NIDA522131 | In rhesus dynamic PET BPND in thalamus, 3–4-fold greater than with 2-[18F]fluoro-A-85380, but at least 8-h recordings required for stable estimation (Chefer et al. 2008) |
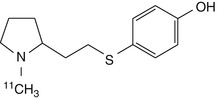 [11C]-SIB-1553A | In rat biodistribution study, 0.5 %ID/g at 10 min and 0.25 %ID/g at 30 min, but no evidence of specific binding discernible against high background (Sobrio et al. 2008) |
17.3.1.1 Nicotine Derivatives
The tracer S-[11C]nicotine, one of the very first positron-emitting receptor ligands (Maziere et al. 1976), was initially developed to investigate the in vivo distribution of nicotine in the context of tobacco addiction, for investigation of diabetes insipidus and for insecticide research (Soloway 1976; Gündisch 2000). With the advent of PET, S-[11C]nicotine was tested for imaging nAChRs in human brain (Nordberg 1993). However, co-administration of unlabelled nicotine failed to displace much of the radioligand, indicating that the PET signal did not sensitively reveal specific binding to α4β2 nAChRs (Nybäck et al. 1994); cerebral S-[11C]nicotine uptake proved mainly to be determined by blood flow, rather than local abundance of nAChRs in vivo (Gündisch 2000).
17.3.1.2 Cytisine Derivatives
The nAChR agonist [3H]cytisine is a useful radioligand for characterisation of α4β2 nAChRs in vitro. However, the cytisine derivatives [11C]ABT-418 and N-[11C]me-thylcytosine (Valette et al. 1997) failed in vivo due to low cerebral uptake and rapid washout. Another derivative, [11C]MPA, showed high-affinity binding (~10–100-fold higher than [11C]ABT-418 and S-[11C]nicotine) in membranes from rat forebrain (Sihver et al. 1998). Furthermore [11C]MPA showed rapid uptake into monkey brain, to a similar extent as that of [11C]ABT-418 and S-[11C]nicotine. Pre-administration of unlabelled S-nicotine (0.02 mg/kg) decreased the peak uptake of [11C]MPA in monkey brain by about 20 %, indicating the presence of some specific binding to nAChRs (Sihver et al. 1999b), but no further studies have been published with this ligand. [18F]fluoropyridinylcytisine was developed as another candidate radioligand for α4β2 nAChR imaging. However, its in vivo distribution in rats did not resemble the regional distribution of nAChRs, and blocking studies with nicotine failed to demonstrate specific binding of this tracer (Roger et al. 2003).
17.3.1.3 Epibatidine Derivatives
Epibatidine has long been known for its high affinity to heteromeric nAChRs (Daly 1998). However it has a rather high toxicity arising from its potency and capacity to activate many different neuronal nAChR subtypes (Avalos et al. 2002). Pharmacological effects of epibatidine related in part to its affinity to the α3β4 nAChR (Tomizawa et al. 2001; Avalos et al. 2002) have limited the application of radiolabelled epibatidine derivatives in human PET studies (Bohnen and Frey 2007). In theory, subtype-specific analogues of epibatidine could present favourable tracer properties with lesser toxicity (Avalos et al. 2002). Although [18F]NFEP ([18F]FPH, norchlorofluoroepibatidine) and [18F]N-Me-NFEP showed good brain uptake and signal-to-background ratios in mouse and baboon brain (Ding et al. 1996; Dolci et al. 1999), their toxicity was considered to be too high for use in man (Horti et al. 1997; Molina et al. 1997; Villemagne et al. 1997; Ding et al. 1999). However, Horti and co-workers successfully synthesised epibatidine derivatives with lesser toxicity, thereby supporting the view that epibatidine could serve as a chemical lead for developing nAChR ligands applicable in PET (Horti et al. 1998a).
Fluorine-18-labelled FPhEP (Roger et al. 2006), a functional antagonist with much reduced toxicity, had faster brain kinetics in baboon than did 2-[18F]fluoro-A-85380 (discussed below), whereas its fluorophenyl analogue [18F]F2PhEP possessed a higher specific binding (Valette et al. 2007), which was quantified as binding potential, which is designated BPND, and is proportional to the ratio B max/K d. However, neither radioligand was adequately displaced by nicotine, which indicated poor specific binding. Patt and co-workers developed [11C]N-methylepibatidine and N-[11C]methylhomoepibatidine (Patt et al. 1999, 2001). The enantiomers of [11C]N-methylepibatidine were compared in vivo in mouse, rat and pig brain. Whereas the (−)-enantiomer showed slower uptake and gradual accumulation in the brain, the (+)-enantiomer showed very rapid uptake and washout. A different mechanism of binding of the enantiomers to nAChRs was proposed as a possible explanation for this difference (Patt et al. 1999). Because of its better kinetics and higher selectivity, [11C]N-methyl(−)epibatidine was investigated in pigs by PET, which showed high brain uptake. However, steady-state binding in thalamus, the region with the highest binding, was not attained within the investigation time (1 h), which may present a significant disadvantage for carbon-11-labelled tracers, due to the 20 min physical half-life. Furthermore high toxicity was observed, precluding its use in humans (Patt et al. 1999). The analogue N-[11C]methylhomoepibatidine had an improved toxicity profile; as was the case with [11C]N-methylepibatidine, the (−)-enantiomer of N-[11C]methylhomoepibatidine showed high uptake in pig brain, while the (+)-enantiomer was rapidly washed out. Although the binding characteristics of N-[11C]methyl-(−)-homoepibatidine in porcine brain suggested suitability for PET imaging studies, its toxicity in mice and rats was comparable to that of N-methylepibatidine, again precluding its use in humans (Patt et al. 2001).
Further development of epibatidine derivatives have focussed on maintaining or improving kinetic profiles while reducing toxicity. One such compound, [11C]2-(6-chloro-5-phenylpyridin-3-yl)-7-methyl-7-azabicyclo[2.2.1]-heptane, showed a thalamus-to-cerebellum ratio of 4.2 at 90 min after injection, indicating high specific binding in rat brain, which was displaceable by pretreatment with nicotine (1 mg/kg). A preliminary PET study of this tracer in a baboon revealed fast brain uptake and a distribution pattern consistent with α4β2 nAChR binding. However binding equilibrium was not reached within 2 h, which is the limit for PET recordings with 11C-labelled radioligands (Huang et al. 2004).
Mu et al. synthesised another series of epibatidine and homoepibatidine analogues (Mu et al. 2006). Of these, the 8-[11C]methyl-8-azabicyclo[3.2.1]octane derivative with a double bond conjugated with the pyridine nucleus, thus restricting free rotation of the pyridine ring, showed high affinity (2 nM) in vitro and at least 100-fold selectivity for α4β4 over α7 nAChRs. Furthermore, it had 50-fold lesser toxicity than epibatidine. Although this radioligand promises to be useful for PET studies, no further experimental data on its evaluation in vivo are available. The antagonistic epibatidine derivative (±)-[11C]NMI-EPB (Ding et al. 2006) had 2.5-fold higher uptake in the baboon brain than did the 3-pyridil ether 2-[18F]fluoro-A-85380, discussed below (Ding et al. 2006). Surprisingly, separation of the (±)-[11C]NMI-EPB enantiomers did not reveal one to be a superior radiotracer: (+)-[11C]NMI-EPB had fast kinetics and low affinity, whereas (−)-[11C]NMI-EPB had slow kinetics (Gao et al. 2007a).
More recently, two further sets of epibatidine analogues (Gao et al. 2007a, b) were developed by the group from Johns Hopkins University, including (−)-[18F]JHU87522 (more recently termed [18F]AZAN) (Gao et al. 2008b; Horti et al. 2010), which displayed promising properties with respect to brain uptake, kinetics, metabolic stability and toxicity. Toxicology and radiation dosimetry experiments in human PET have been reported (Horti et al. 2010). PET studies in baboons confirmed that [18F]AZAN rapidly entered the brain and reached a steady state within 90 min after injection (Kuwabara et al. 2012). Furthermore, blocking experiments with cytisine demonstrated that [18F]AZAN binds specifically with β2-containing (predominantly α4β2) nAChRs, supporting its suitability for nicotinic drug evaluation. Another epibatidine analogue, (−)-[18F]JHU86428 ([18F]XTRA), was proposed, based on its exceptionally high affinity in vitro, as a potential tracer for the less abundant extrathalamic α4β2 nAChRs (Gao et al. 2008b; Horti et al. 2010). An improved radiosynthesis methodology has been reported (Gao et al. 2010), but no further information is available about the ligand’s binding properties in vivo.
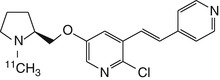
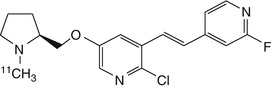
Given that toxicity of epibatidine analogues may be related to high affinity for α3β4 receptors, ligands with a higher selectivity ratio of α4β2 relative to α3β4 are needed. To this end, 18F-labelled stereoisomers of the chloro-fluoro-substituted homoepibatidine analogue, flubatine (previously called NCFHEB), have been synthesised. The two enantiomers of flubatine bind with subnanomolar affinities to membranes from rat thalamus or HEK293 cells expressing the human α4β2 nAChR (Deuther-Conrad et al. 2004). The (+)-enantiomer had twofold higher affinity than did the other stereoisomer. Previous studies have shown that fluoro and norchloro analogues of epibatidine have selectivity for β2-containing receptors (Avalos et al. 2002). Indeed, both enantiomers of flubatine had affinities to α4β2 nAChRs comparable to that of epibatidine, but had 20–60-fold lower affinities to ganglionic α3β4 nAChRs (Deuther-Conrad et al. 2004). The increase in subtype selectivity of flubatine seemingly results in decreased pharmacological side effects compared to epibatidine; injection of 25 μg/kg (+)-flubatine or (−)-flubatine to awake mice was without important pharmacological effects (Deuther-Conrad et al. 2008). The doses expected in a human PET study with [18F]flubatine are more than three orders of magnitude lower (Vaupel et al. 2005), which entails a considerable margin of safety. In addition to its selectivity for α4β2 over α3β4, (−)-flubatine and (+)-flubatine also had considerably better selectivity of α4β2 over α7 receptors than did (−)-epibatidine (Deuther-Conrad et al. 2004).
N-methyl- and N-ethyl-derivatives of flubatine have been synthesised, but displayed lower target affinities and were consequently not considered for radiolabelling (Deuther-Conrad et al. 2004). Similar distribution patterns for (+)-[18F]flubatine and (−)-[18F]flubatine were observed in mice, rat and porcine brain (Brust et al. 2008; Deuther-Conrad et al. 2008; Sabri et al. 2008). Allen et al. provided evidence that nicotine analogues are transported into the brain via the blood–brain barrier (BBB) choline transporter (Allen et al. 2003); this mechanism may also be involved in the brain uptake of epibatidine and homoepibatidine derivatives (Deuther-Conrad et al. 2008). Indeed, addition of flubatine to the incubation medium inhibited the uptake of [3H]choline in immortalised rat brain endothelial cells, which are known to express the blood–brain barrier (BBB) choline transporter (Friedrich et al. 2001), with an IC50 of 370 ± 90 μM (Deuther-Conrad et al. 2008). This result is comparable to the K i of 65 μM obtained for hemicholinium-3 in the same experimental system (Friedrich et al. 2001). Furthermore, in vivo experiments in rats have confirmed the postulated interaction of flubatine with the BBB choline transporter; 50 μM flubatine reduced the transport rate of [3H]choline by 21 %, whereas equimolar epibatidine resulted in a ~40 % reduction (Deuther-Conrad et al. 2008). The stronger interaction of epibatidine is consistent with its higher uptake in mouse brain (London et al. 1995) compared to (+)-[18F]flubatine or (−)-[18F]flubatine.
PET studies in young pigs were performed to compare the brain uptake and kinetics of (+)-[18F]flubatine and (−)-[18F]flubatine with that of 2-[18F]fluoro-A-85380, a 3-pyridal ether discussed in detail below. The brain uptake of both enantiomers proved to be two- to threefold higher than that of 2-[18F]fluoro-A-85380. The binding equilibrium of (−)-[18F]flubatine was reached significantly earlier (~60 min p.i.) than that of the (+)-enantiomer (Brust et al. 2008), consistent with its lesser affinity in vitro. The specific binding of (−)-[18F]flubatine in porcine brain was comparable to that of 2-[18F]fluoro-A-85380, but (+)-[18F]flubatine displayed about twofold higher specific binding. Thus, both [18F]flubatine enantiomers may present advantages over 2-[18F]fluoro-A-85380 for application in human PET studies, especially as pertains to the time to equilibrium binding.
17.3.1.4 3-Pyridyl Ethers
First developed for treatment of Alzheimer’s disease, the 3-pyridyl ethers have long been regarded as promising radioligands for imaging of nAChRs in vivo (Gündisch 2000; Horti and Villemagne 2006; Horti et al. 2010). The 3-pyridyl ethers are equipotent to epibatidine at [3H]cytisine binding sites in the brain, which mainly bind α4β2 nAChRs, but are 100-fold less potent than epibatidine as agonists at α3β4 nAChR receptors (Abreo et al. 1996). This property predicts larger dose margins and minimal cardiovascular or other toxic side effects. The prototype compound A-85380 has similar binding affinities at recombinant α2β2, α3β2 and α4β2 nAChRs in vitro (Xiao and Kellar 2004). In addition, the iodinated derivative 5-[125I]iodo-A-85380 binds with high affinity to α6β2β3 nAChRs in monkey and rat striatum (Kulak et al. 2002). Therefore, 3-pyridyl ethers are properly regarded as β2-selective compounds (Jensen et al. 2005; Lai et al. 2005), furthermore possessing little affinity or efficacy at the α7 nAChR (Sullivan et al. 1996).
As noted above, 2-[18F]fluoro-A-85380 and 6-[18F]fluoro-A-85380 (Horti et al. 1998b, 2000) showed early promise for PET imaging, with a better toxicity profile than epibatidine analogues. Of the two, 6-[18F]fluoro-A-85380 (Scheffel et al. 2000) had superior kinetics, characterised by earlier peak and faster clearance from the brain (Ding et al. 2004), and better target-to-background ratios than were obtained with 2-[18F]fluoro-A-85380 in a comparative study in baboon (Ding et al. 2000a). Although 6-[18F]fluoro-A-85380 has nonetheless not yet found wide use, both derivatives proved successful in human brain imaging (see Sabri et al. in: PET and SPECT in Neurology). The SPECT analogue 5-[123I]iodo-85380 was tested in rhesus monkey (Chefer et al. 1998) and baboon (Fujita et al. 2000). Although hindered by slow kinetics, the radioligand seemed sensitive to changes in the endogenous acetylcholine level (Fujita et al. 2003), a property which will be discussed in some detail below. The methyl-substituted derivative 5-[123I]iodo-A-84543 (Henderson et al. 2004) displayed faster kinetics, but homogenous uptake of radioactivity in baboon brain, in contrast to the spatially heterogeneous pattern of specific binding for the analogue [11C]-A-84543 seen in mouse brain (Kassiou et al. 1998). PET studies in pigs with [11C]-A-186253, a structurally similar tracer, revealed little displacement by cytisine in the thalamus and cortex, indicating excessive non-specific binding (Itier et al. 2004).
The comparably low brain uptake of 2-[18F]fluoro-A-85380 was similar to that previously found with 5-[11C]methyl-A-85380 (2.2 % ID/g brain tissue at 30 min p.i. (Iida et al. 2004)). This may be related to the rather high polarity of these compounds (Zhang et al. 2004), which is regarded as a critical parameter for brain radiotracer uptake (Waterhouse 2003). Nevertheless 2-[18F]fluoro-A-85380 has been successfully used to image nAChRs in non-human primates (Chefer et al. 1999, 2003; Valette et al. 1999; Le Foll et al. 2007) and in rat microPET studies (Vaupel et al. 2007). An optimised radiosynthesis of 2-[18F]fluoro-A-85380 and improved analytical techniques (Mitkovski et al. 2005; Schmaljohann et al. 2005; Kimes et al. 2008) have facilitated its use in human imaging studies (Ellis et al. 2009) (Lotfipour et al. 2012). Despite slow binding kinetics requiring interrupted or continuous PET recordings lasting at least 6 h (Gallezot et al. 2005; Horti and Villemagne 2006), 2-[18F]fluoro-A-85380 remains the most utilised nAChR ligand in human PET studies (see Sabri et al. in: PET and SPECT in Neurology); despite its limitations, it is to some extent the standard against which other PET ligands have been compared.
A series of 5-substituted-6-halogeno derivatives of A-85380 with potentially improved lipophilicity and affinity has been generated (Zhang et al. 2004). PET studies in rhesus monkey with two [18F]-labelled derivatives revealed that their lipophilicity exceeding that of the rather hydrophilic 2-[18F]fluoro-A-85380, resulting in enhanced target-to-background ratios. Imaging studies with another 5-substituted A-85380 derivative, [11C]5-MA (Iida et al. 2004), demonstrated lower total brain uptake and target-to-background ratios than that seen with 2- or 6-[18F]fluoro-A-85380. A further analogue, [18F]ZW-104, is still under investigation (Kozikowski et al. 2005; Valette et al. 2009; Saba et al. 2010). In baboon PET studies, this tracer showed regional radioactivity distribution similar to that of 2-[18F]fluoro-A-85380 and some superior properties, including higher accumulation in the brain, earlier peak uptake in the thalamus and faster washout kinetics. However, it also displayed considerable affinity for α3β2 and α2β2 receptors in vitro (Valette et al. 2009) and rather high non-displaceable (by nicotine) uptake in the striatum, a region with comparably low density of nAChRs.
Many further derivatives were tested in rodent and non-human primate PET studies: [76Br]BAP (Sihver et al. 1999a); [76Br]BrPH (Kassiou et al. 2002); [11C]Me-p-PVC (Brown et al. 2004) and its analogues [11C]JHU85208, [11C]JHU85157 and [11C]JHU85270 (Gao et al. 2007b, 2009); [18F]nifene (Pichika et al. 2006; Easwaramoorthy et al. 2007); and two carbon-11- and fluorine-18-labelled isotopomers of one pyridine-derived ligand. While some of these ligands displayed improved kinetics relative to 2-[18F]fluoro-A-85380, insufficient binding potential or high levels of non-specific binding prevented their further development for human imaging (Easwaramoorthy et al. 2007). Interestingly, specific binding of [18F]nifene could be attributed entirely to α4β2 nAChRs based on studies in β2-knockout mice (Bieszczad et al. 2012). PET imaging of α4β2 nAChRs using the pyridyl ether analogue [18F]nifrolidine has been initiated in rhesus monkey (Chattopadhyay et al. 2005). Although having favourable kinetics, the thalamus-to-cerebellum ratio was lower than that of other α4β2-targeting pyridyl ether analogues (Chattopadhyay et al. 2005), and no further investigations have yet been reported with this compound. Similarly, there have been no follow-up reports on the nifrolidine homologue [18F]nifzetidine (Mukherjee et al. 2004).
The series of pyridyl ether-based compounds [18F]NIDA52189, [18F]NIDA522131 and [18F]NIDA52289 have been synthesised and evaluated by PET in rhesus monkeys (Horti and Villemagne 2006) for imaging of extrathalamic α4β2 nAChRs. Among these, [18F]NIDA52189 (Zhang et al. 2004) and [18F]NIDA522131 (Chefer et al. 2008) were deemed superior to 2-[18F]-FA with respect to extrathalamic binding, but suffered from slow kinetics in vivo.
17.3.1.5 Non-epibatidine- and Non-A-85380-Related Compounds
Carbon-11-labelled Me-p-PVC (Brown et al. 2002) and p-PVP-MEMA, which were selected from the class of (4-pyridinyl)vinylpyridines developed by Abbott Laboratories, possessed picomolar affinity towards α4β2 receptors. Nevertheless, [11C]Me-p-PVC had low binding potential in rhesus monkey thalamus (Brown et al. 2004), and a low target-to-background ratio was obtained in baboon brain for [11C]p-PVP-MEMA in a preliminary PET study (Dollé et al. 2008).
17.3.2 Imaging of Heteromeric β4-Containing nAChR Subtype
The putative β4-selective agonist [11C]-SIB-1553A was assessed by biodistribution and ex vivo brain autoradiography in rats (Sobrio et al. 2008). Its low specific binding suggested a limited potential for further development, although no attempt was made to separate and individually investigate the enantiomers of this racemic radioligand.
17.3.3 Radioligands for α7 nAChRs
Efforts to develop a radiopharmaceutical for PET imaging of α7 nAChR have met with only partial success. The steric and electronic requirements of this site are met by structurally diverse classes of compounds, as reviewed recently (Toyohara et al. 2010b). As noted above, the cerebral expression of α7 nAChR is comparatively low, constituting perhaps one quarter of the density for α4β2 receptors (Spurden et al. 1997; Whiteaker et al. 1999; Hellström-Lindahl and Court 2000). Because of this low natural abundance of α7 sites, high affinity is particularly important for an effective PET tracer. Binding properties of various α7 tracers tested to date are summarised in Table 17.2.
Table 17.2
Results of biodistribution ex vivo and PET imaging studies in vivo of the cerebral binding of α7 nAChR-selective radioligands
Main findings | |
|---|---|
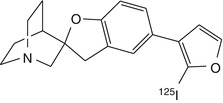 [125I]4 | Biodistribution in CD1 mice showed very limited uptake in the brain and no evidence of displaceable binding (Pomper et al. 2005) |
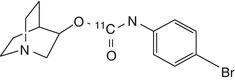 [11C]1 | Biodistribution study in rats showed no regionally selective or specific binding (Dollé et al. 2001b) |
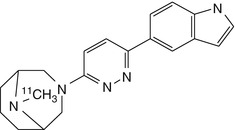 [11C]NS12857 | High uptake in pig brain by dynamic PET, but lack of displacement in vivo (Lehel et al. 2009) |
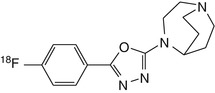 [18F]NS10743 | High uptake in the pig brain by dynamic PET, with clear evidence of displaceable binding in regions of relatively high tracer accumulation (Deuther-Conrad et al. 2011) |
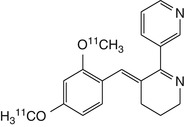 2/4-methoxy-[11C]GTS21 | In baboon dynamic PET, very high initial uptake followed by rapid clearance; relatively little evidence for specific α7 nAChR binding. Brain-penetrating radiometabolites detected in plasma (Kim et al. 2007b) |
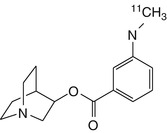 [11C]MeQAA | In rhesus monkey dynamic PET, the R-enantiomer had high cerebral uptake and distribution consistent with α7 nAChRs (Ogawa et al. 2010) |
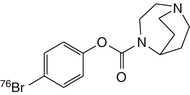 [76Br]SSR180711 | In rhesus monkey dynamic PET, substantial and heterogeneous brain accumulation. Binding globally reduced to level seen in cerebellum by pretreatment with the α7 nAChR agonist SSR180711 (Hashimoto et al. 2008) |
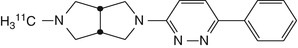 [11C]A-582941 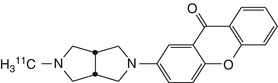 [11C]A-844606 | In rhesus monkey dynamic PET studies, evidence for regional distribution consistent with α7 nAChR expression (Toyohara et al. 2010a) |
 [11C]CHIBA-1001 | In a PET study of one healthy human, evidence for preferential in the hippocampus, cortex and basal ganglia, with slow washout, and least binding in cerebellum (Toyohara et al. 2009) |
17.3.3.1 Quinuclidine-Based Ligands
The lead compound of a series of azabicycle carbamate α7 receptor agonists developed by Astra Laboratories has been labelled with carbon-11 and evaluated in rats (Dollé et al. 2001a). Despite having relatively good brain uptake, no regionally selective or specific binding could be seen. Another series of potential α7-selective imaging agents based on the quinuclidine moiety has been labelled with carbon-11 and iodine-125 (Pomper et al. 2005). Target selectivities of these compounds were modest, and the most affine compounds had significant binding to the 5-HT3 receptor, a structural homologue of α7 nAChR (Zwart et al. 2004).
17.3.3.2 GTS-21
Other potential ligands originate from, for example, benzylidene anabaseine compounds such as GTS-21 (de Fiebre et al. 1995; Meyer et al. 1998). GTS-21 (3-(2,4-dimethoxybenzylidene)-anabaseine) is one of the most promising α7 nAChR agonist drugs currently being evaluated clinically for improving cognition in patients with schizophrenia (Freedman et al. 2008; Tregellas et al. 2011). It has been labelled with iodine-123 (Zhang et al. 2001) and carbon-11 (Kim et al. 2007b). Consistent with the relatively low affinity and specificity of GTS-21 for α7 nAChR, the distribution and kinetics of [5-123I]GTS-21 and [2-11C]GTS-21 in vivo in both baboon and mice were dominated by non-specific binding (Kim et al. 2007a).
17.3.3.3 Diazabicyclononane Derivatives
Recently, the 1,4-diazabicyclo-[3.2.2]nonane skeleton (Bunnelle et al. 2004) has been identified as new structure to improve the α7 receptor-ligand interaction, and the two novel diazabicyclononane-derived PET ligands [76Br]SSR180711 and [11C]CHIBA-1001 were evaluated by PET in conscious non-human primates, with testing in a model of schizophrenia (Hashimoto et al. 2008). Of the two tracers, [11C]CHIBA-1001 demonstrated superior accumulation in the brain, revealing a heterogeneous regional distribution consistent with the localisation of α7 in monkey brain; specific binding was blocked by selective α7 but not α4β2 agonists. Furthermore, it was suggested that this radioligand could be used for measuring occupancy by pharmaceuticals at α7 nAChRs. A first clinical PET study confirmed the suitability of [11C]CHIBA-1001, although the regional binding differences were small in the human brain and α7-specific binding could not be demonstrated (Toyohara et al. 2009). There was no displacement of 30 nM [3H]-CHIBA binding from rat brain membranes by 1 μM α-bungarotoxin in vitro (Tanibuchi et al. 2010). Furthermore, differences in the regional distribution of the binding sites of CHIBA-1001 relative to [125I]α-bungarotoxin were evident in monkey and human brain samples (Tanibuchi et al. 2010). Another rodent study reported low in vitro binding affinity of [3H]-CHIBA and poor in vivo selectivity to α7 nAChRs in rodent brain (Ding et al. 2012).
Among the most promising PET ligands for imaging of cerebral α7 nAChR are the 1,4-diazabicyclo[3.2.2]nonane derivatives developed by NeuroSearch (Peters et al. 2007). Two carbon-11 radioligands were developed, [11C]NS12857 (Lehel et al. 2009) and [11C]NS14492 (Ettrup et al. 2011), along with the fluorine-18 compound [18F]NS10743 (Deuther-Conrad et al. 2009). Evaluation of the latter tracer in mice showed it to permeate readily into brain, obtaining a heterogeneous pattern matching the α7 distribution; this binding was blocked by pre-administration of a selective α7 agonist. The brain uptake of these three 1,4-diazabicyclo[3.2.2]nonane derivatives is generally higher than that of [11C]CHIBA-1001. Although the uptake of [11C]NS12857 was not blocked by pretreatment with α7 nAChR-selective compounds, specific binding was clearly evident for [11C]NS14492 and [18F]NS10743, the two ligands with higher target affinity (Brust et al. 2012).
The general suitability of these derivatives for PET imaging of α7 nAChR was shown by preclinical PET studies in pigs (Lehel et al. 2009; Deuther-Conrad et al. 2011; Ettrup et al. 2011), although the magnitude of BPND, about 0.5, is the absolute minimum for a useful PET tracer. However, in view of the low expression density of α7 nAChR in the brain, a substantial increase in α7 affinity of PET radiotracers may be required for sensitive quantitation (Brust et al. 2012); target affinities of [11C]CHIBA-1001 (K i ~ 35 nM) (Hashimoto et al. 2008; Toyohara et al. 2009) and [18F]NS10743 (K i ~ 10 nM) (Deuther-Conrad et al. 2009) do not predict adequate specific signal in vivo, given the low B max (Koeppe 2001). NS14490, a novel diazabicyclononane derivative, with a K i of 3 nM may be more promising in this regard (Brust and Deuther-Conrad 2012). The distribution of [18F]NS14490 binding in mouse brain autoradiograms correlated with the known pattern of α7 nAChR expression and was displaced with the α7 nAChR ligand methyllycaconitine (Brust and Deuther-Conrad 2012). Very recently, a new highly affine radioligand (K i = 0.3 nM), [18F]DBT10, has been studied in pigs and showed BP values in brain between 2 and 5 (Teodoro et al. 2014, unpublished).
17.3.3.4 [11C]A-582941 and [11C]A-844606
Recently, a new series of octahydropyrrolo[3,4-c]pyrrole derivatives was described by Abbott Laboratories, two of which were selected for labelling with carbon-11 (Toyohara et al. 2010a). Whereas no regional heterogeneity or displaceable binding was evident in mouse brain ex vivo, pretreatment with an α7-specific agonist decreased the total distribution volumes of both tracers in conscious monkey PET studies, indicative of a BPND close to 0.5, as with the NeuroSearch compounds cited above.
17.3.3.5 [125I]I-TSA
A diazabicyclooctane-derived PET ligand with high affinity and selectivity has been radiolabelled and evaluated in mice (Ogawa et al. 2006). Despite a subnanomolar affinity, the high non-specific binding made [125I]I-TSA inadequate for imaging of brain α7 receptors.
17.3.3.6 R-[11C]MeQAA
The two enantiomers of [11C]MeQAA, an azabicyclooctylester-derived compound, were evaluated in mice and conscious monkey PET studies (Ogawa et al. 2009). Although R-[11C]MeQAA showed target-specific accumulation, the in vivo selectivity was insufficient due to binding to the serotonin 5HT3-R.
17.4 Imaging of Neurodegenerative Diseases
Reductions in cortical nAChR binding have been found in patients with diverse forms of dementia, including Alzheimer’s disease, Parkinson’s disease, Lewy body disease, progressive supranuclear palsy and Down’s syndrome (Perry et al. 1986; Picciotto and Zoli 2002). However, there have been few preclinical PET and SPECT studies of nAChRs in animal models of neurodegenerative disease. This is attributable to two considerations: First, models in transgenic mice have only recently become available for some of these diseases while remaining lacking for others, and second, the spatial resolution of small-animal PET and SPECT instruments has until recently been inadequate for regional analysis of neuroreceptors in rodent brain, such that the majority of such investigations have been conducted using autoradio-graphy in vitro. Furthermore, interpretation of preclinical imaging studies are hindered by the occurrence of species differences (Pauly et al. 1989; Quik et al. 2000; Han et al. 2003). Despite these limitations, molecular imaging is emerging as a powerful tool for investigating pathophysiological changes in animal models of neurodegenerative diseases, especially when conducted in conjunction with techniques such as in vivo microdialysis, electrophysiology and histopathology (Higuchi et al. 2012).
17.4.1 Alzheimer’s Disease
A link between cognitive performance and α4β2 nAChR expression in forebrain of healthy rats has been demonstrated in a PET study using the ligand [18F]nifene (Bieszczad et al. 2012), as confirmed by autoradiography ex vivo and in vitro. The three imaging methods showed the same rank order of specific binding by brain region. We anticipate that [18F]nifene PET should allow tracking of dynamic changes in nAChRs during learning acquisition and memory consolidation, in rodents and also in large-brained animals. In contrast to the case for humans (Zanardi et al. 2002), normal aging does not seem to reduce nAChR density in rat brain (Picciotto and Zoli 2002; Schliebs and Arendt 2011).
The hallmark histopathological features of Alzheimer’s disease are extraneuronal amyloid plaques composed of aggregated β-amyloid peptides (Aβ), and intraneuronal neurofibrillary tangles, which are composed largely of hyperphosphorylated forms of tau, a microtubule-associated protein (Thal and Braak 2005). Molecular imaging studies in animal models have primarily targeted fibrillar protein assemblies such as β-amyloid and tau depositions, neuroinflammatory processes and cerebral glucose metabolism (Higuchi et al. 2012). However, these features are closely related to cholinergic hypofunction found in Alzheimer’s disease and relevant animal models (Schliebs and Arendt 2011). Cognitive impairment in Alzheimer’s disease is at least partially associated with loss of cortical nAChRs, which may arise due to toxi-city of soluble β-amyloid (Zanardi et al. 2002; Schliebs 2005; Schliebs and Arendt 2011). Nicotine treatment in a transgenic mouse model (3xTg-AD) can mediate increased tau phosphorylation and decreased β-amyloid load (Rubio et al. 2006).
Impaired cholinergic neurotransmission has been described in the brain of Tg2576 mice, which express the Swedish mutation of human β-amyloid precursor protein (Apelt et al. 2002). Enzyme activities for acetylcholine synthesis (choline acetyltransferase) and degradation (acetylcholine esterase, AChE) did not differ between transgenic mice and non-transgenic littermates. However, a reduction of high-affinity choline uptake and M1-muscarinic receptor density was observed. Autoradiography with [3H]cytisine revealed a significant 20 % loss of α4-containing nAChRs in cingulate and parietal cortices of these animals at an age of 17 months. However, there was no change in the number of basal forebrain cholinergic neurons in the transgenic mice, compared to age-matched wild-type animals.
Evidence for an involvement of α7 nAChR in Alzheimer’s disease was first presented three decades ago (Davies and Feisullin 1981). More recently, a very high-affinity binding of (soluble) β-amyloid to α7 nAChRs has been described in vitro (Wang et al. 2000), supporting the hypothesis that β-amyloid at very low concentrations may initiate neuronal degeneration via an α7 nAChR-mediated inflammatory process (Bencherif and Lippiello 2010). A 20 % reduction in α7 nAChRs labelled with [125I]α-bungarotoxin was evident in the hippocampus, retrosplenial and parietal cortices and thalamus of 3xTg-AD mice at 6 months of age. There was as significant correlation between intraneuronal β-amyloid and reduced α7 nAChR binding in the same mouse model (Oddo et al. 2005). Whereas chronic nicotine administration did not alter α7 nAChR in these mice, there was an increase in α4β2 nAChRs labelled with [125I]epibatidine (Oddo et al. 2005). In contrast to the earlier report, no alteration in α7 nAChR binding was noted in a subsequent study of the triple transgenic 3xTg-AD mice, which closely emulate several features of natural Alzheimer’s disease (Hedberg et al. 2010); this unexpected negative finding was attributed to unknown environmental and/or genetic factors.
The abundance of α7 nAChRs was determined using nanogold-conjugated α-bungarotoxin in the APP(SWE) mouse model of Alzheimer’s disease (Jones et al. 2004). Interestingly, the α7 nAChR binding increased in the transgenic animals until 9 months of age, but had declined at 12 months, most notably in areas of gliosis associated with β-amyloid plaques (Jones et al. 2006). Also [125I]α-bungarotoxin binding decreased in APP(SWE) mice between 9 months of age and 16 months (Hellström-Lindahl et al. 2004).
17.4.2 Parkinson’s Disease and Further Neurodegenerative Diseases
Parkinson’s disease is, after Alzheimer’s disease, the second most common neurodegenerative disorder. The hallmark neuropathology of Parkinson’s disease is selective degeneration of midbrain dopaminergic neurons of the substantia nigra pars compacta (SNpc) and the presence of intra-cytoplasmic inclusions (Lewy bodies) consisting of aggregated α-synuclein (Spillantini et al. 1997) in surviving dopamine neurons. In a 2-[18F]fluoro-A-85380 PET study, the density of α4β2 nAChRs was slightly reduced in the basal ganglia of non-smoking Parkinson’s disease patients (Kas et al. 2009). Another 2-[18F]fluoro-A-85380 PET study of Parkinson’s disease patients reported a widespread reduction of α4β2 nAChR availability in cortical and subcortical regions, which correlated with the severity of mild cognitive or depressive symptoms (Meyer et al. 2009).
Neurotoxins such as MPTP and 6-hydroxydopamine have been used in animal models emulating the nigrostriatal degeneration of Parkinson’s disease (Quik 2004). Lesions of the nigrostriatal pathway in rats reveal a population of [3H]nicotine binding sites on dopamine terminals (Clarke and Pert 1985). More recently, the stimulation of striatal dopamine release by nicotine has been linked specifically to α6β2β3 and α6α4β2β3 nAChRs, which predominate in the basal ganglia (Quik et al. 2011). Quantitative analysis with the α6β2 nAChR subtype ligand [125I]α-conotoxin MII in conjunction with dopamine transport measurements in MPTP-lesioned mice shows an association with presynaptic nigrostriatal terminals (Quik et al. 2003). In that study, much smaller reductions in the binding of [125I]epibatidine (multiple sites) and 5-[125I]iodo-A-85380 (β2-sites) were noted after lesioning, while no change was detected in α7 nAChRs measured with [125I]α-bungarotoxin binding after nigro-striatal lesions. Displacement of [125I]-α-conotoxin MII binding with the analogue E11A was biphasic, allowing resolution of the α6β2β3 and α6α4β2β3 components (Bordia et al. 2007); autoradiographic studies in MPTP-treated mouse and non-human primates, as well as in material from idiopathic Parkinson’s disease patients, revealed the α6α4β2β3 nAChR subtype population to be selectively vulnerable to nigrostriatal damage. Chronic oral nicotine administration was able to protect nicotinic receptors and also dopaminergic markers in MPTP-treated monkeys (Bordia et al. 2006; Quik et al. 2006). There is a current lack of agents for molecular imaging of the particular α6-containing nAChRs subtypes of greatest relevance to Parkinson’s disease.
Huntington’s disease is an autosomal dominant hereditary disorder proceeding to severe cognitive impairment and motor symptoms, notably hyperkinetic involuntary movements (chorea) (Roos 2010). A transgenic rat model of HD, which carries a truncated huntingtin cDNA fragment with 51 CAG repeats under control of the native rat huntingtin promoter, has been developed (von Horsten et al. 2003). Early investigations of nicotinic receptors in post-mortem brain from Huntington’s disease patients did not reveal any significant changes (Perry et al. 1987; Whitehouse and Kellar 1987). However, autoradiographic assessment of 2-year-old transgenic rats revealed significant increase of nAChR in various regions in heterozygous but not homozygous animals (Bauer et al. 2005).
Some forms of epilepsy have recently been associated with alterations of α4 nAChR subtype expression (Raggenbass and Bertrand 2002), and there is experimental evidence that the α7 nAChR may play a role in epileptogenesis (Dobelis et al. 2003). The autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) can be caused by mutations in the neuronal nicotinic acetylcholine receptor (nAChR) subunit genes CHRNA4 and CHRNB2 (Steinlein et al. 1995; Phillips et al. 2001). Relative to age-matched non-smoking subjects, there was a 10–20 % increase in the binding of 2-[18F]fluoro-A-85380 to α4β2 receptors in the brain of patients with autosomal dominant nocturnal frontal lobe epilepsy (Picard et al. 2006); knock-in mice bearing a culprit mutant α4 gene have been prepared (Lipovsek et al. 2008), but have not been investigated by receptor autoradiography or PET.
17.5 Imaging of Stroke and Neuroinflammation
Stroke is the leading cause of adult disability in the United States and Europe and the second leading cause of death worldwide. Stroke is characterised by a loss of brain functions due to rapid disturbances in cerebral blood supply, either as reduced blood flow by thrombosis or embolism (ischemic stroke) or blood leakage (haemorrhagic stroke). Hyperacute mechanisms of stroke-related brain tissue damage, such as excitotoxicity, can be discriminated from delayed factors such as inflammation and apoptosis. Cellular components of the so-called neurovascular unit, which includes neurons, astrocytes and endothelial cells, all express nAChRs (Paulson et al. 2010). Insofar as long-term tobacco smoking is a risk factor for ischemic stroke (Hawkins et al. 2002), it may be relevant that nAChRs were altered in a post-mortem study of smokers; α4 expression was increased in neurons and dendritic processes and α7 expression was decreased in hippocampal neurons and astrocytes (Teaktong et al. 2004). In hypertensive stroke-prone rats, cortical α7 nAChRs are reduced, without concomitant changes in the α4β2 nAChRs (Ferrari et al. 1999). Activation of nAChRs by nicotine promotes endothelial cell proliferation (Villablanca 1998) and leucocyte migration (Yong et al. 1997), which together may increase thrombotic risk. However, nAChR agonism was neuroprotective against excitotoxicity in vitro, an effect which was mediated by growth factors (Belluardo et al. 1998) and also by inactivation of the toxins (O’Neill et al. 2002). These effects were blocked by the α7 nAChR antagonist α-bungarotoxin (Donnelly-Roberts et al. 1996). Interestingly, nicotine increased oedema/infarct size in a rodent stroke model (Paulson et al. 2010). A particular contribution of α7 nAChRs to stroke-related excitotoxicity might reflect their high Ca2+ permeability, especially under depolarising conditions. Treatment with the acetylcholinesterase inhibitor methanesulfonyl fluoride attenuated stroke-induced learning and memory deficits in rats (Borlongan et al. 2005).
These rather discordant findings of effects of nAChR agonism in stroke models may reflect the different contributions of excitotoxicity and also neuroinflammatory processes. The α7 nAChRs expressed on microglia (Shytle et al. 2004) seem particularly posed to mediate inflammatory responses. Nicotinic agonism at these sites suppressed inflammation by decreasing TNF-α production, while nicotine antagonists had the opposite effect (Shytle et al. 2004). In addition to central mechanisms, cholinergic signalling in the “cholinergic anti-inflammatory pathway” involving the vagus nerve may suppress the release of pro-inflammatory cytokines and influence migration T cells from the periphery to brain areas affected by stroke or multiple sclerosis (Borovikova et al. 2000) by a mechanism sensitive to α-bungarotoxin (Pavlov et al. 2003).
Despite this extensive background, there have been very few molecular imaging studies of nAChRs in relevant neuroinflammatory models. We have undertaken PET studies to assess α7 nAChR alterations in the sheep stroke model (Boltze et al. 2008). Recently, we measured [18F]NS10743 binding at 6 months after permanent medial cerebral artery occlusion (pMCAO) by dynamic PET imaging using a clinical tomograph (unpublished data). We found reduced tracer uptake in cortex ipsilateral to the lesion, but increased uptake in the brainstem. The declining α7 nAChR availability doubtless indicates degeneration of neurons in the infarct area, but the distal increases are of uncertain significance. Ongoing studies aim to establish better the time course and histological correlates of the α7 nAChR changes in our stroke model.
17.6 Imaging of Traumatic Brain Injury
Traumatic brain injury is a permanent or temporary impairment of brain functions with an associated diminished or altered state of consciousness caused by external mechanical force. In the western world, it is the most important cause of death in young adults (Olesen and Leonardi 2003). In a human post-mortem study, performed after traumatic brain injury, reduced choline acetyl transferase activity and synaptophysin immunoreactivity in cerebral cortex were found, indicating damage of the cholinergic innervation, but nAChR binding determined by either [3H]nicotine or α-[125I]bungarotoxin was unchanged (Murdoch et al. 1998), suggesting mainly presynaptic mechanisms.
Among various animal models which have been developed to experimentally investigate traumatic brain injury, the fluid-percussion (FPI), weight drop and controlled cortical impact (CCI) models are the most common. The FPI model, which entails transduction of mechanical force to the brain via a fluidic wave (Thompson et al. 2005), causes injuries resembling those observed in the clinic. The CCI model, which was first developed in small animals (Lighthall 1988) and later used in pigs (Duhaime et al. 2000; Alessandri et al. 2003; Manley et al. 2006), entails a piston strike, which results in more focal brain injury than is afforded by the FPI model.
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Tracers for Beta-Amyloid and Other Proteinopathies
 PET Imaging of ABC Transporters in the BBB
PET Imaging of ABC Transporters in the BBB
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 Development of Radioligands for In Vivo Imaging of NMDA Receptors
Development of Radioligands for In Vivo Imaging of NMDA Receptors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


