Prions are a rare cause of human disease but very important to recognize because of their potential for transmissibility and uniformly severe outcome. MR imaging plays an extremely important role in early diagnosis, especially with diffusion-weighted and fluid-attenuated inversion recovery images, which are the most sensitive for depicting prion-induced brain lesions. The lesions are characteristically shown as ribbons of cortical hyperintensity, or basal ganglia or thalamic hyperintensity. The cortical and deep lesions may appear alone or together, and although usually bilateral and symmetric, they may be asymmetric or purely unilateral. When these MRI findings are observed in an appropriate clinical context, the diagnosis of prion disease is very likely.
Prions (proteinaceous infectious particles) are infective agents comprised of protein, lacking nucleic acid. The actual protein, termed cellular prion protein ( PrP C ), is a normal cellular constituent. The infective particle is an isoform of PrP, termed scrapie prion protein ( PrP Sc ), resulting from partial refolding into a beta-sheet configuration, which can then induce further conversion of the normal protein into the infective form. PrP Sc has reduced solubility and partial resistance to proteases, making standard sterilization procedures difficult. Clinically relevant infection occurs in the central nervous system in animals and humans, resulting in varying degrees of spongiform neuronal degeneration, neuronal loss, reactive astrocytic gliosis, and amyloid-like plaques predominantly affecting gray matter structures .
Human forms of prion infection include sporadic, familial (inherited), and acquired. The sporadic forms are divided into sporadic Creutzfeldt-Jakob disease (sCJD) and sporadic fatal insomnia. sCJD is further divided into several subtypes, largely based on which amino acids are encoded by codon 129 in the prion gene. The familial prion diseases consist of familial CJD, fatal familial insomnia, and Gerstmann-Sträussler-Scheinker disease (GSS). The acquired diseases are kuru, iatrogenic CJD, and new variant CJD (vCJD) or “mad cow disease.” Kuru largely disappeared with the end of cannibalism among the Fore people of New Guinea.
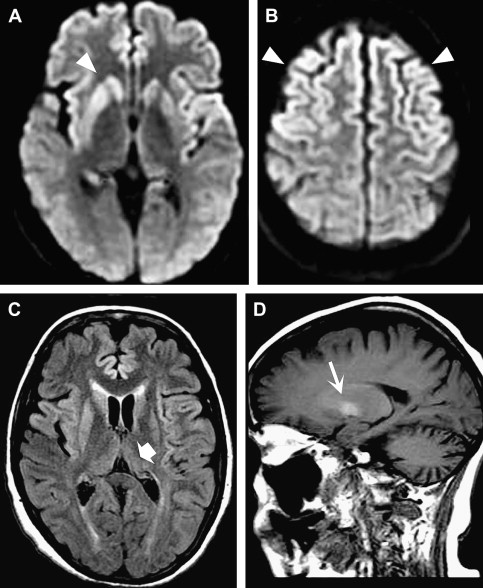
sCJD ( Fig. 1 ) accounts for approximately 85% of human cases, with the remainder being predominantly hereditary, including familial CJD, GSS, and fatal familial insomnia . The hereditary forms are related to mutations associated with different codons of the PrP encoding gene, PRNP . Iatrogenic causes include dura mater grafts, corneal implants, and contaminated human growth hormone . vCJD, which is believed to be transmitted through contaminated meat from cows infected with bovine spongiform encephalopathy, has gained recognition from a public health perspective .
Overall, CJD is rare, with an incidence of less than one per million. The sporadic form is more common among elderly individuals, whereas the variant form is seen in younger patients. sCJD is characterized by rapidly progressive dementia resulting in death over weeks to months. As the disease advances, typical symptoms include the development of motor dysfunction, myoclonus, and, in the later stages, akinetic mutism, with death frequently occurring secondary to respiratory infection. The variant form has prominent psychiatric features early in the disease and a slower progressive course, with a reported median survival of 14 months . According to World Health Organization (WHO) criteria, definitive diagnosis of sCJD is made through histopathologic analysis; however, characteristic periodic sharp wave complexes (PSWC) on electroencephalogram (EEG) or positive cerebral spinal fluid (CSF) 14-3-3 protein assay in the appropriate clinical setting contribute to a probable diagnosis . vCJD does not tend to display typical EEG or CSF findings and has separate diagnostic criteria, in which imaging studies have an important role .
MR imaging is more sensitive than CT and remains the mainstay of imaging investigation. Typical findings of sCJD on conventional sequences include symmetric T2 hyperintensity involving the basal ganglia, in particular the corpus striatum . Less common features include asymmetric striatal involvement and signal abnormality in the thalamus and periaqueductal gray matter . Cerebral cortical signal abnormality, either symmetric or asymmetric, may be seen in the absence of striatal involvement and has become a well-recognized feature, particularly with the greater use of fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted images (DWI) and because these images have greater sensitivity for detecting cortical lesions . Evidence shows that DWI is the most sensitive sequence in CJD evaluation . DWI shows persistent restricted diffusion in the typical areas of involvement that have been reported to progress over the disease course but may normalize in the later stages, possibly related to gliosis . T1 hyperintensity may be seen in the globus pallidi, postulated to be related to prion deposition . Cortical perirolandic sparing has been reported , but subsequent quantitative analysis of apparent diffusion coefficients has shown a similar degree of involvement in these “spared” areas, suggesting that local effects may be masking true regional pathology . MR imaging findings can precede the onset of characteristic clinical disease, particularly with the use of DWI . No mass effect or enhancement is seen, and progression to atrophy occurs in the terminal stages . When only cortical involvement is present, radiologic mimics may include mitochondrial myopathy, encephalopathy, lactic acidosis, and stoke; venous hypertensive disease ; and posterior reversible encephalopathy . With respect to other imaging tests, MR spectroscopy has shown decreased N -acetyl aspartate (NAA), reflecting neuronal loss, but is nonspecific . Positron emission tomography and single photon emission CT (SPECT) have been shown to be sensitive for early changes, with diminished lobar metabolism/perfusion . Although the specificity has not been well established, good correlation between SPECT and DWI has been reported .
In a recent, large, multinational study from 1992 to 2002, MR imaging examination of 1036 patients who had definite sCJD showed characteristic imaging findings in only 40%, which was significantly less sensitive than either EEG (60%) or 14-3-3 CSF assays (90%). However, FLAIR and DWI were not used systematically . Both DWI and FLAIR have greater sensitivity than T2-weighted sequences. Smaller cohorts directly examining DWI have shown greater than 90% sensitivity and specificity for patients who have a definite or probable diagnosis, even in the presence of negative EEG and CSF results , and therefore experts have suggested that it be included in the diagnostic criteria for sCJD .
In a recently published a series of 36 cases, 26 of which underwent DWI imaging, Shiga and colleagues emphasized the value of DWI as an early diagnostic marker for CJD. They compared the percentages of DWI abnormalities, PSWCs on the EEG, detection of CSF 14-3-3 protein, and increase of CSF neuron-specific enolase (25 ng/mL) on the first examination. Disease controls consisted of 32 patients, aged 31 to 84 years, who had progressive dementia or impaired consciousness. DWI showed abnormalities in 92.3% of cases, PSWCs were seen in 50%, 14-3-3 protein was detected in 84%, and neuron-specific enolase increase was seen in 73.3%. Of the 32 control subjects, 2 had false-positive results on DWI. The sensitivity of DWI was 92.3% and specificity was 93.8%. In 17 patients who did not show PSWCs on the first EEG, abnormal DWI findings were still clearly detected. Four patients who had negative results for 14-3-3 protein also showed DWI abnormalities. DWI abnormalities were detected at 3 weeks of symptom duration in four patients in whom PSWCs were not yet evident.
In 26 patients who had CJD, DWI was examined 3 to 25 weeks after onset with a mean duration of 10.7 weeks. Of these patients, 24 showed high-intensity brain lesions on DWI examination, 3 (12.5%) showed lesions only in the caudate heads and putamen, 10 (41.7%) showed linear lesions only in the cerebral cortex, and 11 (45.8%) showed lesions in the basal ganglia and cerebral cortex. Only 3 patients (12.5%) showed lesions in the thalamus. No patients showed high-intensity lesions in the cerebellum. In some cases, the high-intensity lesions detected with sequential DWI did not always progress with the advance of the disease, and in some lesions the signal intensity decreased with disease progression. In some cases, the cortical high signal varied in intensity and anatomic distribution. In the terminal stage with profound brain atrophy, the high-intensity lesions became unclear .
The variant form of Creutzfeldt-Jakob disease
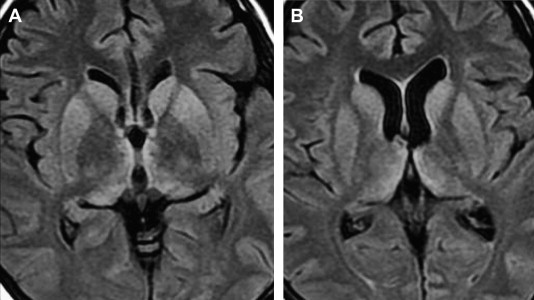
vCJD shows distinctive imaging findings; in particular, symmetric high signal in the pulvinar compared with the signal in the remainder of the basal ganglia. This finding is termed the pulvinar sign ( Fig. 2 ) . The WHO criteria for vCJD includes the pulvinar sign as a component of a probable diagnosis . In a prospective study of 86 patients who had histopathologically confirmed vCJD, symmetric high signal on T2-weighted, proton density–weighted, or FLAIR images was present in more than 90% . The finding was rarely asymmetric. FLAIR also showed dorsomedial thalamic involvement, termed the hockey stick sign , to be positive in more than 90% of patients. Other common areas of abnormality are similar to the sporadic form and include the striatum and periaqueductal gray matter. Less commonly, parietooccipital white matter hyperintensity and cerebral and cerebellar atrophy have been noted . Case reports have described pulvinar signal abnormality in the sporadic form as a potential mimic ; however, experts have noted that true variant cases should have pulvinar involvement greater than the basal ganglia, and that this relative hyperintensity is more specific than simple pulvinar signal abnormality . In other words, pulvinar hyperintensity is an excellent but not pathognomonic indication of vCJD . However, not all of these cases fulfilled the WHO criteria for this sign, which specify that the pulvinar signal must be brighter than the signal in other deep nuclei. In two reports, the WHO criteria were fulfilled, with pulvinar intensity being higher than other deep gray matter nuclei . The pulvinar sign is also seen occasionally with other diseases, such as limbic encephalitis . Apparent diffusion coefficient values may vary within the structures involved .
Iatrogenic prion disease
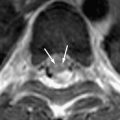
Fewer than 300 cases of human iatrogenic prion disease have been noted worldwide . Most of these infections were caused by cadaveric human growth hormone, dura mater, and corneal grafts. The most frequent MR imaging finding in these cases has been bilateral symmetric hyperintensity of the caudate head and putamen, with the abnormalities appearing earlier on DWI than on T2-weighted imaging . Asymmetric pulvinar hyperintensity has also been reported after a cadaveric dura mater graft . In the single reported case of MR imaging performed on a patient who had a history of corneal grafts, caudate and putamen hyperintensity was found, and FLAIR and DWI showed cortical hyperintensity .
Iatrogenic prion disease
Fewer than 300 cases of human iatrogenic prion disease have been noted worldwide . Most of these infections were caused by cadaveric human growth hormone, dura mater, and corneal grafts. The most frequent MR imaging finding in these cases has been bilateral symmetric hyperintensity of the caudate head and putamen, with the abnormalities appearing earlier on DWI than on T2-weighted imaging . Asymmetric pulvinar hyperintensity has also been reported after a cadaveric dura mater graft . In the single reported case of MR imaging performed on a patient who had a history of corneal grafts, caudate and putamen hyperintensity was found, and FLAIR and DWI showed cortical hyperintensity .
Inherited prion disease
Inherited prion diseases comprise approximately 15% of human cases. Few reports on imaging findings have been published. GSS disease is one of the inherited prion diseases. Clinically, prominent cerebellar ataxia is seen. MR imaging reports on GSS are much less common than with CJD, but these diseases share common MR imaging findings. Most notably, DWI has shown bilateral discontinuous ribbon-like hyperintensities throughout the cerebral cortex and progressive cortical atrophy . Cerebral hypoperfusion on SPECT and diminished NAA may precede findings on conventional MRI . Imaging findings described in other inherited forms include cortical atrophy, cerebellar atrophy, and hypointensity on T2-weighted imaging in the basal ganglia . Some patients have shown no MRI abnormality at all. Some symptomatic patients who had normal MR imaging examinations had 123 I SPECT imaging that showed patchy decreases across the entire cerebrum . In fatal familial insomnia, MR imaging did not show any specific abnormality, or merely mild atrophy . In some presymptomatic carriers, early atrophy was seen .
The Heidenhain variant
Some types of prion disease have been attached with eponymous names because of one or more prominent clinical features. In the Heidenhain variant of sCJD, visual symptoms occur first and persist throughout the disease course. The first change in behavior may be that patients stop reading or watching television because of deteriorating vision. Funduscopic examination usually shows little abnormality. Onset of dementia occurs later. In this clinical setting, MR imaging is extremely valuable because the diagnosis of CJD is rarely suspected. In one series of 11 cases, MR imaging showed symmetric hyperintensities in the basal ganglia in seven patients and marked signal increase in the occipital visual cortex in four. Two cases also showed isolated atrophy in the visual cortex . In another recent series, Cooper and colleagues reported that the Heidenhain variant of CJD accounted for 22 of 594 cases of sCJD. Their patients presented with early, prominent, visual complaints, and in some the only complaints were visual. Of these patients, 77% initially presented to ophthalmologists before consulting any other specialist. Hyperintense cortical ribbons or typical CJD basal ganglia lesions on MR imaging should immediately indicate inclusion of CJD in the differential diagnosis. MR imaging leads to a much more rapid definitive diagnosis than would otherwise be achieved, circumventing the need for many other diagnostic tests ( Fig. 3 ).
Related posts:
 Imaging of Topographic Viral CNS Infections
Imaging of Topographic Viral CNS Infections
 Neuroimaging of Viral Infections in Infants and Young Children
Neuroimaging of Viral Infections in Infants and Young Children
 Imaging of Nonspecific (Nonherpetic) Acute Viral Infections
Imaging of Nonspecific (Nonherpetic) Acute Viral Infections
 Central Nervous System Infections of Herpesvirus Family
Central Nervous System Infections of Herpesvirus Family
 Imaging of Slow Viruses
Imaging of Slow Viruses
 Prion Protein Disease and Neuropathology of Prion Disease
Prion Protein Disease and Neuropathology of Prion Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


