Nucleus
Name
Spin
Frequency (B 0 = 1.5 T)
Inherent sensitivity
Natural abundance (%)
1H
Proton
1/2
63.87
1
99.99
7Li
Lithium
3/2
24.81
0.272
92.58
13C
Carbon
1/2
16.06
0.0159
1.11
19F
Fluorine
1/2
60.08
0.833
100.00
23Na
Sodium
1/2
16.89
0.0925
100.00
31P
Phosphorous
1/2
25.85
0.0663
100.00
35Cl
Chloride
3/2
6.26
0.0047
75.53
39K
Potassium
3/2
2.98
0.00051
93.08
The difference between MRS and MRI is that MRS detects the different chemical components within tissues, whereas MRI shows the anatomical architecture. The targeted nuclei have slightly different resonance frequencies due to interactions via the electrons, which induce small magnetic field change μ e around the nuclei (Fig. 7.1). Because of these small local magnetic field differences, the resonance frequencies of the nuclei will shift known as the “chemical shift.” The information produced by MRS is displayed graphically as a spectrum with a chemical shift (horizontal) axis in parts per million (ppm) and a signal amplitude (vertical) axis enabling identification of chemical components. Since each signal amplitude or area is proportional to each particular metabolite concentration, one can measure relative or absolute values of concentrations of chemicals.

Fig. 7.1
Chemical shift and J-coupling. (a) Since electrons are negatively charged, the electron cloud shielding a nucleus induces an opposing small magnetic moment μ e to the applied field. As a result, the nucleus experiences a slightly different magnetic field (B 0 − μ e), which causes a slight shift of resonance frequency: B = B 0(1 − μ e). The oxygen atoms shift the electron density away from the protons leading to reduced electronic shielding; therefore the resonance frequency of the proton nucleus shifts higher. (b) The proton resonance is split into a doublet due to scalar coupling with the carbon in which the energy level repeatedly switches between high- and low-spin state. This effect is called spin-spin coupling or J-coupling
7.2.1 Chemical Shift
In practice, chemical shift is not expressed in absolute units (such as hertz). This is because the value of chemical shift depends on the applied magnetic field strength; that is, if the magnetic field B 0 is doubled, then the absolute value of the chemical shift will be doubled. Therefore, the chemical shift is measured in relative terms as follows:

In proton MRS, tetramethylsilane (TMS), Si(CH3)4, is traditionally used as universal standard for chemical shift calibration. For example, if the obtained value of the chemical shift between the main peak of N-acetylaspartate (–CH3) and TMS (ω NAA − ω TMS) is measured by 1.5 T scanner as 128 Hz, then the chemical shift will be presented as 128/64,000,000 Hz = 2 ppm. Here, the number of 64,000,000 (64 MHz) is the center resonance frequency (ω 0) of protons at 1.5 T. In case of the resonance frequency of the proton in water, the peak appears at 4.7 ppm.
7.2.2 J-Coupling and Splitting Pattern
The actual local magnetic field encounters an indirect interaction of neighboring magnetic nuclei (spin-spin splitting) causing perturbation of the electron distribution and splitting of NMR signals. This phenomenon, known as spin-spin coupling or J-coupling, provides detailed information on identities of spin-possessing nuclei in a molecule. J-coupling can occur between any species of nuclei with spin, in homonuclear (e.g., 1H–1H) and heteronuclear (e.g., 1H–13C) interactions (Fig. 7.1). Peak splitting from J-coupling shows the same actual frequency values, whatever the externally applied magnetic field strength, so its value is expressed in “Hz” and not in “ppm.” When a species of nucleus is coupled to n equivalent nuclei, its signal splits into an n + 1 multiplet, with component amplitude ratios following Pascal’s triangle: singlet 1; doublet 1:1; triplet 1:2:1; quartet 1:3:3:1; quintet 1:4:6:4:1; etc. Coupling with additional spin-possessing nuclei causes additional splitting of each component of the multiplet, for example, producing a doublet of doublets.
In lactate (CH3–CH(OH)–COOH), for example, the methyl groups (CH3) at 1.31 ppm interact with the methine groups (CH) at 4.10 ppm, and the methyl nucleus signal is split into two equal peaks (a doublet) separated by 6.93 Hz. J-coupling also causes phase evolutions that cause peak and baseline distortions that vary with echo time and field strength [4]. This phenomenon is well known as the observation that the lactate doublet has inverted doublet peaks for TE ≈ 143 ms (Fig. 7.2). Since the overlapping within individual metabolites and between other metabolites due to J-coupling cancels each other by dephasing effect at longer echo times, metabolites such as γ-aminobutyric acid glutamate and glutamine are hard to measure using long echo times (TE > 50 ms) with in vivo proton spectroscopy [4].

Fig. 7.2
MR spectrum of lactate showing different phase modulations caused by J-evolution for different echo times. The proton signal from the methyl group (CH3) is split into a doublet by the proton of the methine group (CH). On the other hand, the proton signal from the methine group (CH) is split into a quartet with a signal ratio of 1:3:3:1 by the three protons of the methyl group (CH3). In coupled spin systems, the amplitude and phase of the resonance are modulated and suffer large signal attenuation during T2 decay
7.3 Metabolites Seen by Proton MRS in the Brain
At high magnetic fields of 7 T and above, at least 18 metabolites can be identified noninvasively by MRS [5, 6]. In clinical MR scanners at 1.5 T, the metabolites distinguishable in proton MRS are limited to several metabolites such as choline-containing compounds, creatine + phosphocreatine, glutamine + glutamate, myoinositol, and N-acetylaspartate + N-acetylaspartylglutamate (NAAG). γ-Aminobutyric acid (GABA) can be measured too by a spectral editing method like MEGA-PRESS in a clinical MR machine at 3.0 T [7].
7.3.1 N-Acetylaspartate (NAA)
NAA is an amino acid present in the brain in high concentrations, second in amount to glutamic acid. Interestingly, NAA is found only in the central and peripheral nervous systems and is thought to reflect neuronal density and dysfunction. The NAA concentrations in the brains of mammals and birds are especially high (up to 10 mM), while in the brains of amphibians, reptiles, and invertebrates, it is much lower [8, 9]. The NAA concentration in the peripheral nervous system and the retina is about 10–20% that of the levels in the central nervous system [10–12]. NAA shows a conspicuous singlet peak at 2.02 from the N-acetyl (CH3–CO–NH–) moiety in vivo MRS and has attracted intense interesting from researchers, but little is known about its metabolism and functions. Several functions have been suggested to explain the presence and amount of NAA in the nervous system, including the following:
- 1.
NAA works as an osmoregulator/molecular water pump, removing metabolic water from neurons [13].
- 2.
It is an immediate precursor for NAAG synthesis.
- 3.
It shuttles acetate into oligodendrocytes for myelin synthesis.
- 4.
It is a storage and transport form of acetyl-coenzyme A (acetyl-CoA) in the nervous system that lacks substantial glycogen reserves [14]. Acetyl-CoA cannot cross cell membranes, so it must be converted to citrate in the Krebs cycle for transport to the cytoplasm. NAA may function as an exportable molecule carrying both acetate and aspartate.
NAA is synthesized from l-aspartate and acetyl-CoA by the enzyme aspartate n-aspartate transferase (Asp-NAT) in an oxygen- and ADP-dependent manner. The synthesis increased with age within the first few years of life, and plateaus from age 2 to 20 years, depending on the brain region [15, 16]. In the adult brain, NAA synthesis occurs almost only in neurons [17]. NAA synthesis is coupled to glucose metabolism, and the metabolic relationships can be shown in the following equation [18, 19].

The time required for NAA synthesis is comparable to that for the brain glycogen synthesis from glucose in the awake rat [20], which takes about 10–14 h [21, 22], and much slower than the synthesis of glutamate, around 1–2 h [23–25]. Furthermore, the NAA synthesis rate in the human brain is in the same range as that found in rat cortex [22]. Therefore, the estimated complete turnover of NAA from glucose is thought to be very slow, taking approximately 70 h [21]. Asp-NAT has not been identified molecularly due to the difficulty of purification, but a recent study indicates that N-acetyltransferase 8-like protein (NAT8L) is a strong candidate for Asp-NAT [14, 26]. It has long been believed that NAA is synthesized within mitochondria, but NAT8L is localized to the membrane of endoplasmic reticulum [27]. Glutamate acts as a substrate for NAT8L, with a 50 times lower affinity, and shows competitive inhibition of the aspartate incorporation into NAA [26].
NAA is released to extracellular fluid space and taken up by glial cells via sodium-dependent dicarboxylate (NaDC3) transporters [28]. In oligodendrocytes, NAA is hydrolyzed by aspartoacylase (ASPA) to aspartate and acetate [18], and that acetate is used in lipid synthesis [29] (Fig. 7.3). Deficiency of ASPA causes leukodystrophy, which appears as spongy degeneration of the white matter that is known as Canavan disease [30]. In this case, MRS shows a high concentration of NAA which is fairly specific to this disease. Interestingly, there was one reported case of NAA and NAAG deficiency in the brain, possibly due to lack of Asp-NAT [26], and the patient suffered lack of expressive speech, truncal ataxia, seizures, mental retardation, and microcephaly [31–33].

Fig. 7.3
N-Acetylaspartate (NAA) and N-acetylaspartylglutamate (NAAG) metabolism and function of the brain. Aspartate (Asp) is synthesized in the mitochondria and exported by neuronal AGC1 ARALAR. NAA is synthesized from acetyl-coenzyme A (Ac-CoA) and Asp by aspartate-N-acetyltransferase (Asp-NAT), now thought to be a membrane-bound microsomal enzyme. Ac-CoA arises from citrate (Cit) through ATP citrate lyase and is transported into cytoplasm from mitochondria through the citrate carrier. The NAA transporter NaDC3 is primarily expressed in astrocytes and oligodendrocytes and transports NAA into these cells in a Na+-coupled manner. NAA carries Ac-CoA from neurons to oligodendrocytes for synthesis of myelin lipid. Aspartoacylase (ASPA) cleaves NAA giving rise to acetate and Asp. NAAG is synthesized by NAAG synthetase (NAAGS) in an ATP-dependent reaction. NAAG is released from presynaptic terminals as a neurotransmitter. Released NAAG is degraded into NAA and glutamate extracellularly by GCP-II. NAAG also binds to mGluR3 on presynaptic membranes and inhibits release of neurotransmitters, including glutamate and GABA. NAAG activates mGluR3 on astrocytes, initiating astrocyte Ca2+ waves responsible for astrocyte-astrocyte and astrocyte-vascular system signaling and induction of vasodilation and consequent hyperemia in locally activated neurons
7.3.2 N-Acetylaspartylglutamate (NAAG)
NAAG is the most abundant dipeptide of the brain and is synthesized by NAAG synthetase catalyzing the ATP-dependent condensation of NAA and glutamate [34]. Absolute measurement of NAAG has shown that its concentrations in the brain are approximately 0.6–1.5 mM in gray matter and 1.5–2.7 mM in white matter. NAAG is released together with glutamate during neuronal depolarization. NAAG activates metabotropic glutamate receptor mGluR3 [35], localized on presynaptic site of neuron terminal and on astrocyte. The presynaptic mGluR3 receptors inhibit release of neurotransmitters, including glutamate and GABA [36–40]. NAAG also acts on NMDAR as a partial antagonist. These negative modulatory effects of NAAG must be important in synaptic plasticity and also in neuronal protection against excessive glutamate release. NAAG activates mGluR3 on astrocytes, initiating astrocyte Ca2+ waves responsible for astrocyte-astrocyte and astrocyte-vascular system signaling and induction of vasodilation and consequent hyperemia in locally activated neurons [19]. As NAAG is a signal molecule specifically targeting the astrocytic mGluR3 receptor, its triggering of brain-activation-related focal BOLD response is thought to be stronger than that of glutamate [41] (Fig. 7.3).
NAAG is hydrolyzed into NAA and glutamate by glutamate carboxypeptidase type II (GCP-II) which is also known as prostate-specific membrane antigen (PSMA) or folate hydrolase 1 (FOLH1), and the products are transported into astrocytes and oligodendrocytes. GCP-II inhibitors have been shown to reduce brain glutamate resulting in neuronal protection in preclinical models of stroke, amyotrophic lateral sclerosis, and neuropathic pain [42].
7.3.3 Creatine and Phosphocreatine (Cr)
Creatine and phosphocreatine (PCr) are abundant in metabolically active tissues such as the muscle, heart, and brain. PCr anaerobically donates a phosphate group to ADP to form ATP via creatine kinase during the first few seconds following an intense muscular or neuronal effort, by the well-known pathway as follows:

Creatine kinase is highly concentrated in the cerebellum, choroid plexus, and hippocampal granular and pyramidal cells [43]. In postmortem examination, brain-type creatine kinase levels were found to be decreased in the patients with AD, especially in the temporal, cingulate, and occipital cortices [44]. Creatine is biosynthesized endogenously by two sequential steps. The first step is guanidinoacetate synthesis from arginine by l-arginine/glycine amidinotransferase (AGAT) expressed mostly in the kidneys, and the second step is creatine synthesis from guanidinoacetate by guanidinoacetate methyltransferase (GAMT), expressed mostly in the liver. Creatine is imported through the blood-brain barrier by a specific transporter, SLC6A8, also called creatine transporter (CRT), expressed in the microcapillary endothelial cells. CRT is widely expressed in the adult mammalian brain, particularly in regions related to limbic functions [45–47]. In general, half of creatine stores originate from endogenous synthesis, while the other half is supplied from food. However in the brain, CRT is scarce in astrocytes, particularly in their feet lining microcapillary endothelial cells [48]. Thus the transport of creatine from blood to brain seems to be relatively insufficient [49, 50], and the brain depends more on its own creatine synthesis through AGAT and GAMT expression than on external supply from blood [51, 52]. Both AGAT and GAMT are found in all types of brain cells but rarely together in any one cell [48, 53–55], so guanidinoacetate must be moving from cells holding AGAT to cells holding GAMT [52]. However, guanidinoacetate is not transported without CRT, so in case of CRT deficiency, even with AGAT and GAMT expression in the CNS, MRS finds little or no Cr (creatine and PCr) in the brain [56]. GAMT is expressed strongly in oligodendrocytes, moderately in astrocytes, and very weakly in neurons [55], suggesting that the final step of creatine synthesis in the brain predominates in glial cells.
In proton MRS, the singlet peaks at 3.07 and 3.96 ppm arise from the methyl and methylene protons of creatine and PCr, respectively. The resonances of creatine and PCr are very close, but the methylene resonance of creatine at 3.931 ppm and PCr at 3.930 ppm can be separated into two components at magnetic fields of 7.0 T and higher. Total creatine (Cr) level seems to be fairly stable with time unless there is some severe disease, which is why Cr is usually reliable as an internal standard. However, the Cr signal may increase or decrease in disease.
7.3.4 Choline-Containing Compounds (Cho)
There are several types of phospholipids, including phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), sphingomyelin, phosphatidylinositol, and cardiolipin. Of these, only PS carries a net negative charge. Newly synthesized lipids are incorporated into the plasma membrane, where PC ends up mainly on the external surface, whereas the aminophospholipids, PS and PE, end up mostly on the cytoplasmic [57]. In most eukaryotic membranes, PC and PE make up some 60–85% of the phospholipid fraction, but the relative amounts of other phospholipids vary between species and between cell types [58]. The major pathway of biosynthesis of PC and PE is the cytidine diphosphate (CDP)-choline and CDP-ethanolamine pathway, known as the “Kennedy” pathway (Fig. 7.4). PE is converted to PC by phosphatidylethanolamine N-methyltransferase (PEMT), which occurs mostly in the liver in mammals. Phospholipids are hydrolyzed by phospholipases. PC is catabolized into lyso-PC by phospholipase A or into glycerophosphocholine (GPC) by phospholipase B. Lyso-PC is rapidly converted to GPC by lysophospholipase (LPL). GPC is then catalyzed into free choline and glycerol-3-phosphate (G3P) by choline phosphodiesterase or phospholipase D (PLD) (Fig. 7.4).

Fig. 7.4
Pathways of glycerophospholipid biosynthesis and catabolism. The glycerophospholipid is synthesized by three enzymatic steps. First, choline kinase (CK) catalyzes the phosphorylation of choline with ATP to form phosphocholine. The second step is the synthesis of high-energy donor CDP-choline, which is rate-limiting of the pathway. This step is catabolized by the phosphocholine cytidylyltransferase (CCT) to synthesize CDP-choline from phosphocholine and CTP. The final step is the synthesis of phosphatidylcholine, which is catabolized by the cholinephosphotransferase (CPT) using CDP-choline and a lipid anchor such as diacylglycerol (DAG) or alkyl-acylglycerol (AAG) to form phosphatidylcholine [172]. The synthesis of water-soluble glycerophosphocholine (GPC) could involve sequential activity of a phospholipase A (PLA) and a lysophospholipase (LPL) or, alternatively, activity of a single phospholipase B (PLB). GPC is then degraded by hydrolysis to glycerol-3-phosphate (G3P) and choline, catalyzed by phospholipase D (PLD). The synthesis and catabolism of phosphatidylethanolamine is similar to the process of phosphatidylcholine. The MRS-visible choline-containing compounds (*) usually present in cytosol as choline, phosphocholine, and GPC. GPC is produced by cell membrane breakdown, but phosphocholine is produced both in catabolic and anabolic process. EK ethanolamine kinase, ECT phosphoethanolamine cytidylyltransferase, EPT ethanolaminephosphotransferase, PEMT phosphatidylethanolamine N-methyltransferase
Most of the choline in the brain is located in cell membranes, bound as phospholipids, and this immobile choline is invisible by MRS. Therefore, the Cho peak of MRS reflects water-soluble forms of choline such as phosphorylcholine and GPC. The contribution of free choline seems to be very little because its concentration is usually under detection limit of MRS. The concentration of the total amount of these MRS-visible choline-containing compounds in human brain tissue is approximately 1–2 mM [59–61]. Choline, phosphorylcholine, and GPC show a strong singlet resonance at 3.22 ppm from protons of trimethyl moieties (–N(CH3)3) at 3.2 ppm, whereby small differences can be observed between 3.5 and 4.3 ppm in multiplet resonances from the remaining CH2 protons. The concentration of PE in human brain is approximately 0.5–1.0 mN [62], and it can be detected by MRS, as it shows multiplet resonances around 3.1–3.2 and 3.8–4.0 ppm. Possible overlap of resonances from ethanolamine, mIns, glucose, and taurine should be taken into account when using short echo MRS.
The Cho peak is considered as a marker of cellular density and membrane turnover. Elevated Cho signals have been found in tumors, in active demyelination sites, and in inflammation. There is a report that the Cho peak is increased in AD, and in patients with amnestic type MCI (aMCI) who progress to AD, but decreased in patients with aMCI who remain stable [63]. It has also been shown that a higher Cho/Cr ratio in the white matter is associated with an increased risk of developing AD within 4 years [64]. It is reported that soluble Aβ oligomers can open cell channels, letting calcium ions into the cell leading to mitochondrial dysfunction, inflammation, and even cell death [65, 66]. Low concentrations of soluble Aβ1–40 or Aβ1–42 peptide induce calcium-dependent release of arachidonic acid via inducing PLA2 activation in a calcium-independent manner [67].
7.3.5 Myoinositol (mIns)
Myoinositol is a major osmolyte released from brain cells, mainly from astrocytes, and also serves as the precursor to phosphatidylinositols. Phosphatidylinositol 4,5-bisphosphate (PIP2) is the source of the inositol 1,4,5-triphosphate (IP3), which is a second messenger in signal transduction and lipid signaling. Inositol is a cyclic sugar alcohol consisting of six-carbon rings with six NMR detectable protons and exists in nine possible stereoisomers, of which mIns is the most abundant in human tissue, representing about 90% of total inositol content. The concentration in the brain is normally 4–8 mM, sufficient for MRS sensitivity. NMR of mIns shows a doublet of doublets centered at 3.52 ppm from 1CH and 3CH protons and a triplet at 3.61 ppm from 4CH and 6CH protons. In practice, these proton peaks are seen as a singlet at about 3.57 ppm at 1.5 T. The proton resonances from 4CH and 6CH partially overlap with the glutamate and glutamine multiplet peaks. Since the T2 relaxation time of the 4CH and 6CH protons is short due to their strong coupling, mIns can only be detected at short echo times. The second most concentrated isomer is scyllo-inositol (0.2 mM), which possesses six symmetric protons and evokes a singlet at 3.34 ppm.
Myoinositol is obtained from food and by de novo synthesis. Synthesis and breakdown occur predominantly in the kidneys, so both neurons and glia need to take it in via transporters, sodium/myoinositol cotransporter SMIT1 and SMIT2 and H+/myoinositol cotransporter HMIT. SMIT is expressed in the kidneys and in the brain [68, 69] and is found in both neural and non-neural cells [70]. It is well known that the upregulation of SMIT1 and SMIT2 occurs under hyperosmotic conditions [71–75], whereas no such osmoregulatory function has been reported for HMIT in mammals. SMIT activity is increased by extracellular alkalization, although under physiological conditions, this effect is small. HMIT is activated by extracellular acidification and is close to inactive at pH 7.5 or higher [76]. Since the activity of HMIT is regulated by pH, mIns uptake may induced at any site of low extracellular pH caused by synapse activation or activated glial cells around the synapse [76]. However, the buffering system in the brain is very complicated, and little is known about HMIT. There are many conflicting findings on HMIT, for example, that it is predominately expressed in astrocytes [76] or specifically in neurons [77] and is localized on cell surfaces [78] or in intra-cytosolic membranes [77, 79, 80] and regulating intracellular signaling.
There are several clinical reports of decreased or increased mIns levels of the brain when measured by MRS. It is known that neurodegenerative disorders such as AD, Huntington’s disease [81], and spinocerebellar ataxia [82] show increased level of mIns in the affected brain areas. People with Down syndrome show significantly increased mIns concentrations in their parietal [83] and hippocampal [84] regions before neuronal degeneration becomes apparent [85]. Interestingly, the human SMIT gene is located in q22.1 on chromosome 21, and inability to downregulate expression of three copies of the osmoregulatory gene could result in increased flux of mIns [86]. Chronic hyponatremia may cause reduction in mIns [87]; and conversely, chronic hypernatremia increases brain mIns concentration [88]. Ammonia is normally converted to urea in the liver, but in cases of chronic liver dysfunction, accumulated ammonia in the brain is detoxified in astrocytes by conversion to glutamine because of the lack of the urea cycle in the brain. This may increase intracellular osmolarity, and astrocytes may compensate by releasing mIns [89]. Lithium treatment may reduce mIns levels in bipolar patients [90, 91]. Valproate causes 20% reduction of mIns level in mouse frontal cortex [92]. These drugs may inhibit monophosphatase, an enzyme required for inositol recycling and de novo synthesis [93]. However, the proportion of mIns participating the phosphatidylinositol second messenger cycle system appears to be low to affect total brain mIns signal.
Whereas mIns has long been seen as a marker of glial cell proliferation, based on the fact that higher mIns levels are found in cultured astrocytes than in neurons in vitro [94, 95], the true mechanisms of increase or decrease of its concentration remain largely unclear. Brain mIns levels may not always correlate with other molecular markers of gliosis [96–98]. It should be noted that high concentrations of mIns are observed in some types of cultured neurons [99, 100] and that mIns is actively taken up into most types of mature brain cells, including neurons and glia [70]. Therefore the assumption that mIns is a specific glial maker should be treated with caution.
7.3.6 Glutamine and Glutamate (Glx)
In mammalian brains, glutamate is both the main excitatory neurotransmitter and the direct precursor of the inhibitory neurotransmitter γ-aminobutyric acid (GABA). The concentration of glutamate of the brain is about 6–12 mM, with a significant difference between gray and white matter. Under steady-state conditions, two thirds of the glucose oxidation in astrocytes is used for de novo synthesis of glutamate [101], and most astrocytic glutamate is converted to glutamine that is transferred into glutamatergic neurons, completing the glutamate-glutamine cycle. The uptake of synaptic glutamate provides up to 80% of the substrate for glutamine synthesis [102]. Since glutamate transport by excitatory amino acid transporter (EAAT) is coupled to and driven by cotransport of sodium, proton, and potassium ions, any increase in rate of the cycle is matched by increased energy consumption. In this model of glutamate metabolism, glutamate metabolism in the brain is compartmentalized into two pools: a larger neuronal pool comprising about 80% of total glutamate, metabolized with relatively slower turnover time, and a smaller astrocytic pool comprising about 20% of total glutamate, which is rapidly converted to glutamine [103].
NMR of glutamate shows a doublet of doublets centered at 3.75 ppm from the single 2CH methine proton, while the resonances from the other four protons appear as multiplets between 2.04 and 2.35 ppm. Glutamine and glutamate are structurally similar, and their resonances overlap each other. The 2CH methine proton of glutamine shows triplet peak at 3.75 ppm, while the multiplets peaks from the methylene protons are closely resonated between 2.12 and 2.46 ppm. Glutamine has also two amide protons at 6.82 and 7.73 ppm. Unfortunately it is difficult to distinguish between glutamate and glutamine by MRS at low magnetic field strength, so the total amount of glutamate and glutamine is often summed up as Glx. There are minor contributions from glutathione and GABA to this combined signal. Since the concentration of glutamine is almost the same as that of glutamate in the brain [104, 105], the glutamine signal is the major confounder of glutamate quantification. The strong coupling of protons also causes different appearances of spectrum at different echo times and different magnetic field strengths. These factors make their observation and quantification difficult with MRS, although the sum of glutamine and glutamate (Glx) can be quantified with high accuracy. Since the glutamine and glutamate levels can change in opposite directions, the total amount of these metabolites tends to be stable. Most clinical MRS studies can measure only this combined amount, resulting in a reduced sensitivity of Glx as a disease marker. Moreover, about 21% of the total glutamate of the brain is thought to be located in intracellular compartments, in which NMR hardly detects it [106].
7.4 Spectrum Analyses
The area under a peak is proportional to both the number of nuclei per molecule of a metabolite resonating at that frequency and the metabolite concentration to which the nuclei belong. Therefore, quantitative measurement can be done either from the amplitude of the first data point of the time domain signal, that is, free induction decay (FID), or from the determination of peak areas in the frequency domain after Fourier transformation of FID. Reliable metabolite quantification by MRS is still challenging. One important factor causing systematic error is the baseline correction. The baseline in any MR spectrum is much affected by field inhomogeneities, tissue heterogeneities, and patient movements, all of which may distort the line shape. There have been various proposals to correct these unwanted distortions during signal processing [107–112]. For accurate quantification in short echo MRS of the brain, correction must be made for the contribution of macromolecules to the baseline [113, 114].
A number of advanced techniques based on mathematical models have been developed for analyses of MR spectra. Time domain methods are grossly divided into “black-box” and interactive model methods. HSVD and HSLVD are commonly used black-box methods, so as VARPRO and AMARES are well-known interactive methods. Frequency domain methods can be also divided into nonparametric and parametric methods. Nonparametric quantification is performed by simple integration of the areas under the peaks of interest, but this method cannot disentangle overlapping peaks, and its accuracy is highly dependent on proper phase correction, which is far from trivial [115]. The parametric methods are based on a model function to fit the spectrum represented by LCModel.
VARPRO is a basic program for time domain analysis that uses a simple Osborne’s Levenberg-Marquardt algorithm for local optimization. It has been replaced by AMARES, which allows accurate and efficient parameter estimation of MRS signals, using a sophisticated nonlinear fitting in the time domain with prior knowledge about the signal parameters, the model function (Lorentzian, Gaussian, Voigt model), and the type of signal (FID or echo). These methods cannot remove nuisance peaks such as macromolecules, lipids, unsuppressed water, and other unknown signals.
Two other programs, AQSES [109] and QUEST [116], make use of a metabolite basis set, which can be built up from simulated spectra from quantum mechanics such as NMR-SCOPE [117] or GAMMA [118] or from in vitro spectra [119]. AQSES [109] is a program that has been especially developed for short echo MRS. In its framework, VARPRO is modified to enable prior knowledge of upper and lower bounds on the nonlinear parameters, equal phase corrections for each metabolite, and optimize the nonlinear least squares problem. AQSES is freely available as Java open-source software AQSES GUI [109, 118], as a measurement system in the Matlab® graphical user interface SPID [120], and as a plug-in in the jMRUI software package (version 4.1) [121].
The jMRUI software [122] runs in either the time or the frequency domain, using signal processing algorithms such as VARPRO, HLSVD, and AMARES [123]. Basis spectrum fitting in jMRUI is performed by the Quantum ESTimation (QUEST) [116] algorithm, which fits the data to a basis set in the time domain. Both AMARES and QUEST enable handling of macromolecule signals and spectral baselines in a number of ways by truncation or down-weighting of the initial points in FID. jMRUI can also use basic methods such as HSVD or HSLVD that can be applied to remove a residual water signal as preconditioning.
LCModel (Linear Combination of Model) is widely used commercial software [124] that uses frequency domain fitting by a linear combination of individual peak profiles of metabolites. The complexities of metabolite spectra become an advantage for this program, since overlaid peaks from different metabolites can be separated from the information at the peaks of other metabolites. This software is fully automatic (noninteractive) and operator independent, so it is highly suitable for studies across centers. LCModel uses a nearly model-free lineshape function and presumes neither a Lorentzian nor a Gaussian lineshape model. It uses the water signal as an internal reference and integrates with the basis set contained in the software, acquired from the standard phantom. It uses the Levenberg-Marquardt nonlinear fitting algorithm and gives some error assessments of the Cramér-Rao type or the like. Mosconi et al. [125] recently compared those four measurement methods (LCModel, AMARES, QUEST, and AQSES) to see whether they produced different statistical results and found that LCModel worked best in their simulation.
7.5 Quality Control
Reliability of fit can be estimated by Cramér-Rao lower bounds (CRLB) [126]. CRLB gives the minimal possible variance on a fit parameter, so any peak measurement with this value over 20% is usually judged as unreliable. MRS is highly sensitive to inhomogeneities in the magnetic field, so shimming is an important procedure for spectral quality. Shimming reduces field inhomogeneity reading to improve water suppression, signal-to-noise ratio (SNR), and spectral resolution. Since SNR is directly reflected in the CRLB that is more directly linked to confidence limits, CRLB seems to be a better parameter by which to judge the quality of quantified data than mere statements of SNR. Linewidth is defined as the full-width at half-maximum (FWHM) peak height in the frequency domain and also reflected in the CRLB indirectly. Broad linewidth means poor spectral resolution that is critical for model fitting. One should note that CRLB indicates only the quality of the spectral fit and does not necessarily reflect the quality of the original data. CRLB is calculated on the assumption that the fitting model and prior knowledge are correct and that systematic errors are not reflected. Possible sources of systematic error are inaccurate T1 and T2 values for each metabolite, incorrect prior knowledge, difficulties in reference-setting and calibration, ROI position or size, structural mixture of GM/WM/CSF, outer volume signal contamination, and operational mistakes [127]. Fit residual is the difference between the measured signal and the model fit, and ideally this should be flat or only white noise if the model fitting is perfect. Fit residual is usually checked by visual inspection, but numerical estimation has been worked out [128]. In LCModel, residues are used as noise level of SNR. Kreis [127] recommended the following acceptance criteria for MR spectral data as follows: FWHM <0.1 ppm (6.4 Hz at 1.5 T, 12.8 Hz at 3 T), CRLB ≦ 50%, the fitting residual should not contain unexplained features, and the spectra should not contain artifacts. From our experience, it is better to set CRLB acceptance criteria as <15% for clinical use regarding possible dementia.
7.6 Acquisition Parameters and Sequences for MRS
The range of echo time (TE) in brain MRS varies from 18 ms to 288 ms, affecting signal peak height and shape depending on the spin-spin relaxation time (T2) and J-coupling of each metabolite. Since diphase and signal loss of protons occurs more rapidly in metabolites with short T2 than long T2, the height of metabolite peaks will change with TE in MRS. For example, in case of the relation between Cr and NAA, since the T2 of Cr is significantly shorter than that of NAA, the peak ratio of NAA/Cr becomes larger in the spectrum obtained from long TE than short TE. Since the T2 relaxation times for Glx and mIns are very short, the peaks of these metabolites cannot be detected at long echo times (Fig. 7.5). This is more obvious in Cho/Cr ratio because T2 for Cho is longer than that for NAA. The T2 values of the metabolites also differ depending on the environment, even in the difference between gray and white matter. Longer echo time is sometimes used for lactate identification. Lactate and lipid resonances are overlaid together, but the lipid resonances may decay fully in case of long echo times that facilitate identification of the lactate peak.

Fig. 7.5
MR spectra of posterior cingulate cortices obtained from normal elderly volunteer. The peak ratio of NAA + NAAG/Cr is 1.266 at echo time = 30 ms, whereas it is 1.784 at echo time = 144. LCModel calculates each metabolite concentration by using tissue water as an internal standard. It should be noted that the default of LCModel for T2 correction of water (T2 = 80 ms) is set at TE = 30 ms, i.e., exp.(−TE/T2) = exp.(−30/80). Therefore if researchers use any TE other than 30 ms, the concentration value of LCModel must be corrected. If TE = 144, then the metabolite concentrations (TE = 144) = (values of LCModel) × exp.(−144/80)/exp.(−30/80) = (values of LCModel) × 0.243. For about NAA + NAAG, it should be 16.893 × 0.243 = 4.105 mM that is too low when compared to the value of 6.416 mM obtained from the results at TE = 30 ms. This is because of the effects of T2 relaxation of NAA and NAAG in vivo, which are uncertain and might differ in each individual. Therefore it is better to use long TR and short TE for quantitative measurement of metabolites to minimize the uncertain effect of T1 and T2 relaxation of the metabolites in vivo
Theoretically, metabolite signals are more affected by uncertain T2 when using long TE than short TE that will cause errors in absolute quantification (Fig. 7.5). Therefore using shorter TE seems to be more profitable for absolute quantification. The other advantage of using short TE is to gain higher signal intensity and to enable detection of metabolites with short T2 such as mIns, glutamate, glutamine, GABA, scyllo-inositol, and lipids. On the other hand, this may cause unwanted distortion of the spectrum baseline and superimposition of peaks from metabolites that may lead to errors in both absolute and relative metabolite quantitation. At short TE, multiple resonance peaks between 2.05 and 2.45 ppm from glutamine, glutamate, and GABA partially overlap the NAA peak at 2.01 ppm. Since the information contained in the short TE and long TE spectra are actually complementary, it may be desirable to use short and long TE sequences as far as permitted by total scanning time, patient tolerance, and cost.
There are two major sequences for volume localization in single-volume MRS: stimulated echo acquisition mode (STEAM) and point-resolved spectroscopy (PRESS). STEAM offers advantages for using MRS of very short TE and more effective water suppression than PRESS because the pulses of water suppression can be applied prior to the first slice selective pulse and during the TM phase [129]. The advantages of PRESS are that it gives double the signal intensity given by STEAM, it is less disturbed by motion and by diffusion, and it does not suffer from multiple-quantum effects. STEAM has a tendency to acquire a volume larger than the chosen VOI, whereas PRESS has the opposite tendency [130].
The methods for absolute quantification of metabolites are summarized elsewhere [131]. In brief, there are three main methods: internal water reference, external reference, and electrical reference. In the method using internal water reference, the signal from unsuppressed tissue water from the same VOI for MRS is used as internal standard. Researchers need to correct the results by %CSF of the VOI and by T1 and T2 relaxation times of each metabolite and water. The inaccuracy of the method is caused by uncertain tissue water content, uncertain T1 and T2 relaxation times of each metabolite, receiver gain instability, and compartmentation such as gray and white matter in the VOI.
7.7 Clinical Application of MRS for Dementia
Many kinds of disorders cause dementia including neurodegenerative disease, vascular or small vessel disease, brain trauma, hydrocephalus, inflammatory diseases, and metabolic disorders. Of these, neurodegenerative diseases represented by Alzheimer’s disease (AD), dementia with Lewy bodies (DLB), and frontotemporal dementia (FTD) are frequently encountered. Recent studies have been disclosing the causes of neurodegenerative dementia at molecular levels, and disease classification is becoming better understood in terms of pathogenesis (Table 7.2).
Table 7.2
Classification of degenerative dementias
Pathology | Disease | Tau | Parkinsonism |
|---|---|---|---|
Amyloid β | AD | 4R + 3R(n) | − |
α-Synuclein | DLB | 4R + 3R(n)a | ++ |
Tau | SD-NFT | 4R + 3R(n) | − |
AGD | 4R(n,g) | − | |
PSP | 4R(n,g) | + | |
CBD | 4R(n,g) | + | |
FTDP-17 | 3R,3R + 4R,4R | + | |
Pick | 3R(n,g) | − | |
TDP-43 | FTLD-U (PPA, SD) | − | |
FTLD-MND(ALS) | − |
The common features seen by MRS in those degenerative disorders are decreased concentrations of NAA or NAA/Cr ratio and increased concentration of mIns or mIns/Cr ratio. Moreover, these findings show a tendency to region specificity, for example, to the hippocampus and the posterior cingulate and precuneal cortices (PCC) in cases of AD, to the occipital lobes of DLB, and to the frontal lobes in case of FTD. Therefore MRS has potential clinical utility to differentiate those disorders. However, searching for such regional distribution anomalies of metabolites may not make MRS the optimal choice in this context, even if using multi-voxel technique. There are many other examinations that can be more adequate for differential diagnosis, such as FDG PET, CBF SPECT, and MRI morphometry. Moreover, new techniques of molecular image targeting pathognomonic biomarkers have been developed, for example, Pittsburg Compound-B (PiB) PET for beta amyloid, PBB3 PET for tau, imaging of dopamine transporter (DAT) or metaiodobenzylguanidine (MIBG) for DLB, and PK11195 PET for microglia. Molecular imaging with target-specific ligands is a promising approach to the early diagnosis of dementia and the evaluation of anti-dementia therapy. Given such alternative diagnostic tools, how can MRS be best exploited for clinical diagnosis of dementia? There is no clear consensus on this. However, there are several clues from previous studies. Firstly, NAA decrease and mIns increase can be identified by MRS before morphometrical change becomes significant. Secondly, MRS provides additional information to clinicians, which improves diagnostic accuracy (Fig. 7.6). MRS is noninvasive, without use of radioactive agents, and is uniquely able to provide information on cerebral metabolites that may reflect neuronal or glial activity or transmission. Moreover, MRI and MRS can share the examination time, which is cost-effective and expeditious.

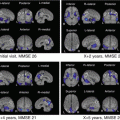
Fig. 7.6
Illustrative case of Alzheimer’s disease. A 56-year-old female visited our hospital with her family having a complaint of memory disturbance. She had come to ask the same questions repeatedly following her retirement at 1 year before. At screening, her MMSE score was 28, and CBF SPECT did not show any remarkable abnormality. The hippocampal volume estimated by VSRAD showed slight atrophy (z score = 1.36), which was equivocal as to diagnosis. However, her verbal recognition score in the WMS-R was under 50, and her logical memory II score was 0, indicating deep cognitive impairment. MRS showed significant decrease of NAA + NAAG concentration under 2SD (our institutional range [151]) in her right and left hippocampi and posterior cingulate gyrus. She had no parkinsonian symptoms, and MIBG SPECT showed normal appearance of the heart-to-mediastinum uptake ratio (H/M ratio)
7.7.1 Age-Related Change
Findings on the brain metabolites changes related to normal aging have been inconsistent. Brooks et al. reported that the absolute concentration of NAA decreased significantly with age (r = −0.42) with an overall decrease of 12% between the third and seventh decades, whereas any decrease of the concentrations of Cr and Cho was not significant [132]. However, some recent papers showed rather different results. In one study revised from their previous study [133] by adding correction of transverse relaxation time and percentage of cerebrospinal fluid of the voxels centered in the frontal gray and white matter, parietal gray matter, and basal ganglia, the concentration of NAA and Cho showed no significant age-related change, but Cr concentration was significantly increased when measuring frontal white matter [134]. This paper also showed the significant age-related declines in glutamate, especially in the parietal gray matter and basal ganglia [134]. Similar results were reported measuring spectrum of the centrum semiovale, which showed that Cr concentration increased with age (r = 0.495, p < 0.001), whereas there were no age-related changes in NAA, Cho, and mIns [135]. Another paper [136] showed significant decrease in NAA concentration (r = −0.36, p = 0.007) and increase in mIns concentration (r = 0.37, p = 0.007) in the supraventricular white matter. The Cr and Cho concentrations were unchanged with age, but Glx showed U-shaped change with higher concentrations in the young and old subjects [136].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


