Etiology
Langerhans cell histiocytosis (LCH) is an uncommon disease. Several synonyms, including histiocytosis X, eosinophilic granuloma, and Langerhans cell granulomatosis, have been used in the past, but the term Langerhans cell histiocytosis is now preferred. The previously used term histiocytosis X included several entities with similar morphologic but different clinical manifestations. These entities included Letterer-Siwe disease, which is often fatal and occurs in children and infants with multiorgan involvement ; Hand-Schüller-Christian disease, which has a more indolent clinical course in children and adolescents and is characterized by lytic skull lesions, diabetes insipidus, and exophthalmos ; and eosinophilic granuloma, a disease of adults with lung and bone involvement. The nomenclature of these various entities has been simplified, and LCH is now basically subdivided into single-organ or multiorgan involvement. Pulmonary involvement in LCH usually occurs in isolation but also is found in most adult cases with a multisystem disorder.
Although viruses such as Epstein-Barr virus, papillomaviruses, and herpesviruses have been previously implicated as a cause of LCH, evidence from molecular biology studies has not substantiated the link. However, the association of pulmonary LCH with cigarette smoking is strong, with supporting evidence from both clinical and pathologic studies. The exception is pulmonary LCH in children, which often occurs as part of a systemic disorder and is not associated with cigarette smoking. Isolated pulmonary LCH in adults is almost exclusively encountered in smokers (>90%), with some studies reporting almost 100% of affected individuals being smokers.
Supporting evidence for the association of LCH and smoking includes the fact that the earliest histopathologic finding in pulmonary LCH is a peribronchiolar interstitial infiltrate, a similar distribution to that seen in smoke-induced small airways disease. In animal studies a Langerhans-like interstitial granulomatous reaction was seen to resolve when exposure to cigarette smoke was withdrawn. Additionally, when compared with nonsmokers, smokers display an increased number of Langerhans cells in bronchoalveolar lavage fluid. Despite these findings, the exact mechanism of the role of cigarette smoking in the pathogenesis of the condition is unclear.
A rare familial association has been reported, with an affected relative occurring in less than 1% of cases in one large multicenter study showing only 1 of 274 adults (0.4%) as having familial LCH. Further evidence for a genetic predisposition is provided by chromosomal instability in peripheral lymphocytes in LCH.
Prevalence and Epidemiology
Despite the strong association with smoking, pulmonary LCH remains an uncommon disease, and accurate data of incidence and prevalence are problematic. With the widespread use of high-resolution CT today such surgical lung biopsy is performed much less frequently, and the diagnosis often is made based on a combination of clinical and imaging findings. In a study in which histopathologic confirmation was not obtained in all patients and diagnoses were made based on clinical and high-resolution CT appearances, pulmonary LCH was diagnosed in 6.6% of 1382 cases of diffuse interstitial lung disease. Previously, pulmonary LCH was described as more common in men, but more recently it has been described in women, and currently the sex incidence is thought to be approximately equal, which may reflect the increasing number of female smokers. It occurs more commonly in whites than in other ethnic groups, and most individuals are in their 20s or 30s, with a mean age at onset of symptoms of 33 years and mean age at diagnosis of 35 years in a large multicenter study.
Clinical Presentation
Patients usually present with cough and dyspnea. Other symptoms may include fever, malaise, and chest pain; however, in many patients the symptoms are mild and nonspecific. Up to 25% of individuals may be asymptomatic with radiographically apparent disease. Approximately 15% of patients may present initially with pneumothorax. In one study of 102 patients with pulmonary LCH, 16% had pneumothorax and for those patients, the age at the time of initial diagnosis was significantly younger (median, 29 years) than in patients without pneumothorax (median, 41 years). A significant proportion (60%) of patients may have recurrent pneumothorax. Hemoptysis rarely may occur. Bone lesions may cause chest wall pain and tenderness. Physical examination frequently is normal, and finger clubbing is rare.
In contrast to the pediatric form of the disease, multiple organ sites are less often affected but have been described, including pituitary, skin, eye, colon, heart, lymph nodes, and brain. In a large study multiple organ involvement occurred in only 28% of adult patients. In all cases with single-organ and multiorgan involvement, the most common affected site was the lungs (58%).
Pathophysiology
Pathology
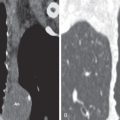
Pulmonary LCH is characterized by histiocyte proliferation. Langerhans cells occur in the normal lung ; however, the histiocytic cells in LCH have distinguishing morphologic and immunohistochemical features, suggesting “activated” Langerhans cells ( Figs. 33.1 and 33.2 ). Most studies have described clonal proliferation of histiocytes secondary to an abnormal inflammatory immunologic response. Activated Langerhans cells are poorly susceptible to apoptosis, or programmed cell death, which may result in repeated stimulation of T cells.


At a microscopic level, a cellular infiltrate is centered around the membranous bronchioles in the early stages of pulmonary LCH. The interstitium becomes involved in more extensive disease, with varying proportions of Langerhans cells, eosinophils, lymphocytes, and fibroblasts, and a characteristic stellate appearance may occur as a result of central fibrosis. Although the stellate pattern is typical, a more symmetric nodular cellular infiltrate without central fibrosis, which may be related to an airway, also can be seen. Occasionally, intraalveolar buds of granulation tissue may give an appearance similar to organizing pneumonia, although this appearance is relatively uncommon. A “desquamative interstitial pneumonia–like reaction” is a frequent histopathologic accompaniment to LCH resulting from pigmented macrophages infiltrating the surrounding airspaces. Respiratory bronchiolitis also may be seen, supporting the hypothesis that smoking-related interstitial lung disease is a spectrum with overlapping features.
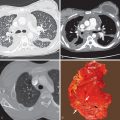
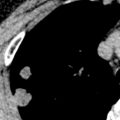
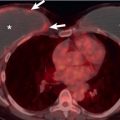
The cystic spaces seen on macroscopic specimens ( Fig. 33.3 ) and imaging seem to result from destruction of the bronchiolar wall and progressive enlargement of the airway lumen, with surrounding fibrous tissue. It has been hypothesized that an increase in cyst size may occur as a result of a “ball-valve” effect secondary to partially obstructed bronchioles. Concurrent emphysema is a frequent finding that is not surprising given the association with smoking. The lesions coalesce as the disease becomes more advanced, with increasing fibrosis, emphysema, and parenchymal destruction. In end-stage pulmonary LCH, the lung may be almost entirely replaced by fibrosis and cystic spaces. Pulmonary vascular involvement is a feature of late-stage LCH with luminal obliteration, intimal fibrosis, and medial hypertrophy. Progression of these features may result in pulmonary arterial hypertension, which may complicate end-stage pulmonary LCH.

Several studies have correlated histopathologic features with high-resolution CT findings. In early disease centrilobular nodules on CT correlate histopathologically with florid granuloma formation. In contrast, cysts on high-resolution CT correlate with cavitary granulomas, cysts with fibrous walls, or a combination, and an association between cysts and air-trapping has been reported. The presence of ground-glass opacities on high-resolution CT correlates with respiratory bronchiolitis/desquamative interstitial pneumonia–like changes. In advanced disease high-resolution CT may show large cysts and architectural distortion, with granulomas still seen on histopathologic specimens consistent with ongoing disease activity.
Lung Function
In early disease, despite the presence of radiologic abnormalities, many patients may have normal lung function; however, the most common pulmonary function abnormality is a reduction in diffusing capacity for carbon monoxide (DLCO). The reduction in DLCO is likely due to a combination of a pulmonary vasculopathy and parenchymal disease. In advanced disease restrictive, obstructive, and mixed pulmonary function abnormalities have been described. Obstruction may be due to bronchiolar narrowing secondary to peribronchiolar inflammation and fibrosis or coexisting emphysema. There is good correlation between the high-resolution CT appearances, including quantification of disease extent with objective measures, and functional parameters in pulmonary LCH.
Manifestations of the Disease
Radiography
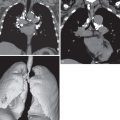
In early disease the radiograph may be normal, with abnormalities seen only on CT. The lung volumes are characteristically preserved or enlarged. If disease is identifiable on radiography, the initial features are of a nodular or reticulonodular pattern with an upper and middle zone distribution that characteristically spares the costophrenic angles ( Fig. 33.4 ). Less commonly, significant involvement of the lower lobes occurs, but even in these cases the costophrenic angles are generally spared. The distribution of nodules and curvilinear or reticular opacities is symmetric without central or peripheral predilection ( Fig. 33.5 ). As disease progresses a reticular pattern occurs from a composite of superimposed multiple cysts ( Fig. 33.6 ). Because of the evolving pattern from nodules to reticular abnormality/cysts, a combination of these features is frequently seen (see Fig. 33.5 ).



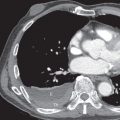
As previously noted, pneumothorax is a common complicating radiographic feature. Uncommon radiographic features include mediastinal lymphadenopathy, consolidation, pleural effusion, and a solitary pulmonary nodule. In advanced disease there may be large areas of destroyed lung with parenchymal distortion and hyperinflation, and distinguishing pulmonary LCH from advanced emphysema may be impossible ( Fig. 33.7 ). Pulmonary arterial hypertension may result in enlargement of the pulmonary arteries in advanced disease and is associated with a negative impact on survival ( Fig. 33.8 ).