Idiopathic Pulmonary Arterial Hypertension
Idiopathic pulmonary arterial hypertension (IPAH) represents pulmonary arterial hypertension in its purest form. By definition, IPAH exists when an underlying cause of the pulmonary arterial hypertension (PAH) cannot be identified. It is a rare disease with a poor prognosis and is characterized by luminal obliteration of small pulmonary arteries. The overall result is increased resistance to pulmonary blood flow, increasing pulmonary artery pressure (PAP), and ultimately, right ventricular failure and death.
Women are affected more commonly than men, with a female-to-male ratio of approximately 1.7:1. Cruelly, there is a predilection for IPAH to affect otherwise normal young women of childbearing age. The mean age for developing the condition is approximately 40 years, but it can occur at any age. Elderly patients often have other coexisting cardiac and respiratory disease, making the diagnosis of pure IPAH more challenging.
Most patients with IPAH present with exertional dyspnea, developing over months or years. This classical, although nonspecific, symptom is thought to be due to the inability of the right heart to raise output on exertion. Chest pain, syncope, and peripheral edema are more common in advanced IPAH and indicate right ventricular failure.1,2
The clinical signs of IPAH include right ventricular heave, loud pulmonary component of the second heart sound, a pansystolic murmur of tricuspid regurgitation, and a right ventricular third sound. Jugular venous distension, hepatomegaly, peripheral edema, ascites, and cold extremities indicate patients in a more advanced state with right ventricular failure at rest. Central cyanosis may also be present in advanced cases.
Unfortunately, the absence of findings on clinical examination and nonspecific symptoms frequently lead to a delay in referral to the appropriate specialist center and, subsequently, a delay in diagnosis and treatment. Symptoms are often initially attributed to poor physical fitness, especially in overweight patients, and the diagnosis only becomes apparent with the development of chest pain, syncope, or edema.
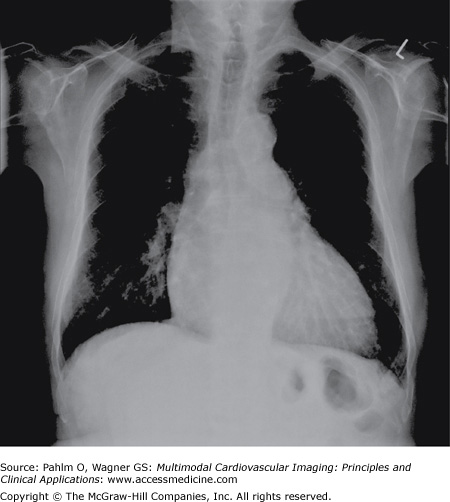
The chest radiograph may give the first clue to the presence of IPAH, providing invaluable information on the heart size, the lung parenchyma, and the pulmonary vasculature.
The main chest radiograph features of IPAH consist of enlargement of the main pulmonary arteries and rapid tapering of the vessels as they extend to the periphery of the lungs, giving rise to peripheral oligemia. The heart, particularly the right-sided chamber, is usually enlarged but may be of normal size (Fig. 24–1).
In IPAH, the ventilation/perfusion scan is usually normal or shows heterogeneous perfusion.3 Occasionally, larger mismatch perfusion defects may be seen in IPAH, thought to be as a result of thrombosis in situ.
The key features of IPAH on computed tomography (CT) are as follows:
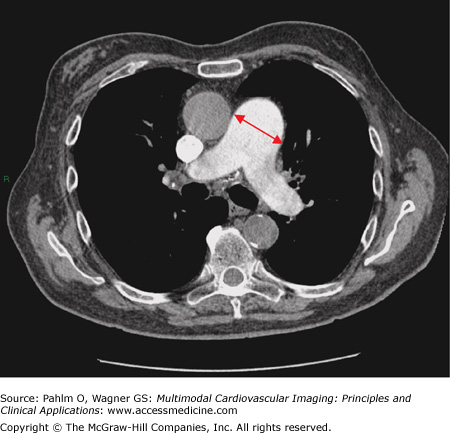
Enlargement of the main pulmonary artery (Fig. 24–2). In one study, a main pulmonary artery diameter of ≥29 mm had a positive predictive value of >95% for PAH.4
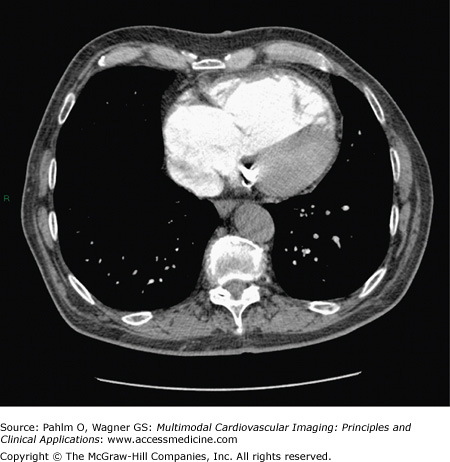
Right ventricular dilatation and hypertrophy and enlargement of the right atrium (Fig. 24–3). Flattening or “bowing” of the interventricular septum can also be observed.
Reflux of contrast from the right atrium into the inferior vena cava when CT pulmonary angiogram is performed. This occurs as a result of tricuspid regurgitation, and the extent of reflux correlates with mean PAP; however, it can be observed in normal patients.5
The presence of pericardial effusions.
Small centrilobular ground-glass nodules.
Bowing of the interventricular septum towards the left ventricle.
Play Video
The electrocardiogram (ECG) may demonstrate right ventricular hypertrophy and strain and right atrial dilatation in IPAH.
Pulmonary function tests in IPAH often reveal normal spirometry but reduced diffusion capacity for carbon monoxide.
The use of echocardiography in the assessment of IPAH focuses on detection of elevated PAP, evaluation of the right ventricle, and exclusion of other possible differential diagnoses.
In IPAH, chronic progressive pressure loading results in right ventricular (RV) remodeling, particularly RV hypertrophy and later RV dilatation. RV dilatation can result in tricuspid annular dilatation, often causing significant tricuspid regurgitation. As IPAH progresses, there is impaired RV diastolic function and increased RV end-diastolic pressure, with displacement or “bowing” of the interventricular septum toward the left ventricle.
PAH can be suggested by abnormalities on a number of echocardiographic views:
Parasternal long axis view: The RV will appear dilated (Fig. 24–4), and in severe cases, there is compression of the left ventricular cavity with “bowing” of the interventricular septum.
Short axis view: The RV is again dilated, with the characteristic D-shaped left ventricle visible (Fig. 24–5).
Apical four-chamber view: There is dilatation and hypertrophy of the RV and often hypertrophy of the moderator band (Fig. 24–6). Tricuspid regurgitation jets will be visible in the majority of patients with pulmonary hypertension, and application of continuous wave Doppler mapping on this jet allows calculation of the tricuspid regurgitation velocity (Fig. 24–7). This velocity represents the RV to right atrial pressure difference and can be used to estimate pulmonary artery systolic pressure (PASP) through the Bernoulli equation: PASP = RV systolic pressure = 4 (VTR)2 + RAP, where VTR is tricuspid regurgitant velocity and RAP is right atrial pressure.
Tricuspid annular plane systolic excursion (TAPSE): TAPSE represents the basal to apical shortening of the RV during systole. TAPSE will be reduced when there is impaired RV systolic function, and a TAPSE of less than 1.5 cm is associated with poor prognosis in PAH.6
A pericardial effusion may be visible on a number of views, and the size of the effusion has prognostic importance in IPAH.
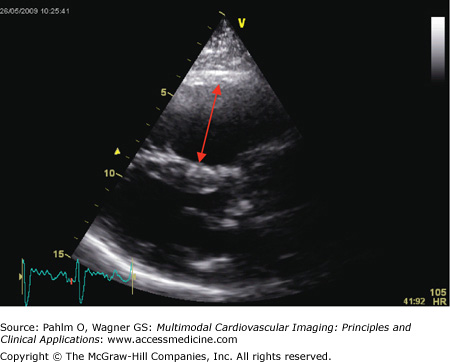
Figure 24–5.
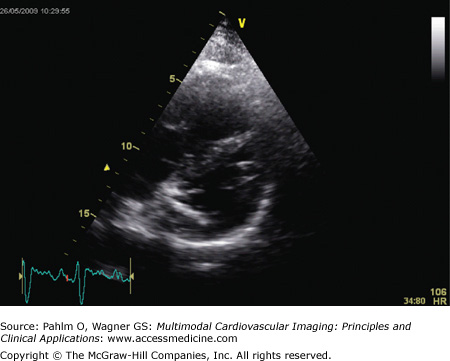
Echocardiogram. Short axis view of same patient with idiopathic pulmonary arterial hypertension as seen in Fig. 24–4. There is right ventricular dilatation and D-shaping of the left ventricle
Figure 24–6.
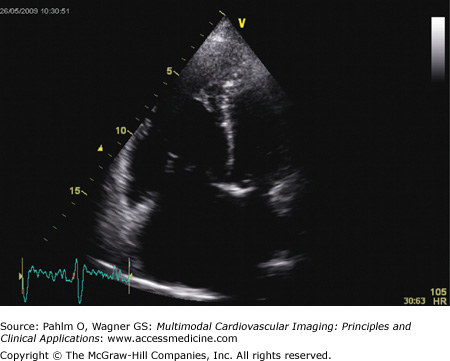
Echocardiogram. Apical chamber view of patient with idiopathic pulmonary arterial hypertension seen in Figs. 24–4 and 24–5. There is marked dilatation of the right-sided cardiac chambers, with interventricular septal bowing.
Figure 24–7.
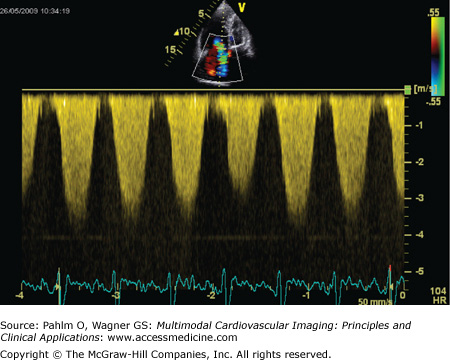
Application of continuous wave Doppler mapping to the tricuspid regurgitant jet observed in Fig. 24–6. The tricuspid regurgitation velocity is approximately 4 m/s, giving a pulmonary artery systolic pressure of 64 mm Hg + right atrial pressure.
Cardiac magnetic resonance (CMR) imaging is ideally suited for examining the complex geometry of the RV. Improvements in CMR technology have reduced the length of breath-holds required to obtain high-quality images, particularly important in patients with IPAH who are often dyspneic on minimal exertion.7
A diagnosis of IPAH is supported the following CMR abnormalities:
- Increased RV end-diastolic volume (RVEDV)
- Reduced RV ejection fraction, stroke volume, and cardiac output
- Poor left ventricular filling, observed as a low left ventricular end-diastolic volume (LVEDV)
- Bowing of the interventricular septum toward the left ventricle (Fig. 24–8)
- RV hypertrophy, occurring as a result of the increased pulmonary afterload (Fig. 24–9)
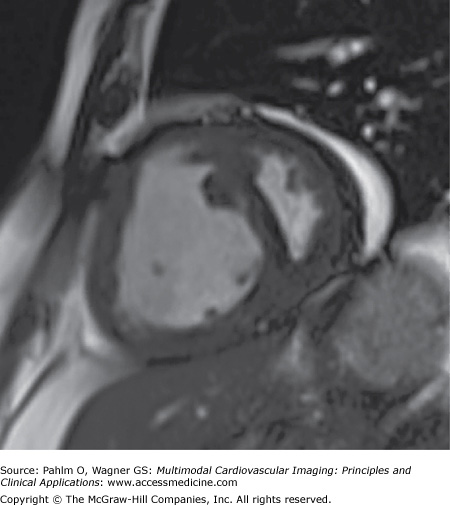
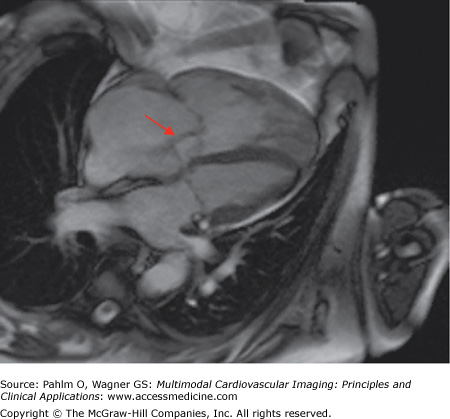
RV hypertrophy can be assessed using ventricular mass index (VMI), which is the ratio of RV mass to LV mass. VMI has been shown to correlate closely with mean PAP obtained at right heart catheterization.8 Studies have also shown that an increased RVEDV, poor left ventricular filling, and low stroke volume are associated with poor prognosis in IPAH.9
Velocity-encoded CMR imaging is another approach for assessing the patient with suspected IPAH. This technique allows us to quantify blood flow within the main pulmonary artery and calculate peak and average pulmonary artery flow velocities and provides an alternative method for estimating stroke volume and cardiac output. Pulmonary artery distensibility can also be measured by CMR and has been found to be significantly lower in PAH patients compared with normal subjects.10
Right heart catheterization remains the gold standard for the diagnosis of IPAH. Patients typically have a markedly elevated mean PAP, usually ≥50 mm Hg (normal, <25 mm Hg). A low cardiac output and high pulmonary vascular resistance (PVR) indicate severe disease and poorer prognosis. If PAP is high, then vasoreactivity testing in an expert center should be done because a successful response mandates different therapy.
There is as yet no cure for IPAH, so management is usually divided into general measures, disease-targeted therapy, and surgical intervention.11,12
- Oxygen: Acute oxygen therapy can improve pulmonary hemodynamics in hypoxic and normoxic patients. Patients with resting partial pressure of oxygen in arterial blood (PaO2) of <8 kPa may be prescribed oxygen for at least 15 h/d.
- Anticoagulation: IPAH is associated with abnormalities in coagulation and fibrinolytic pathways and impaired platelet function. Anticoagulation may prevent vascular thrombotic lesions and pulmonary embolism.13,14
- Diuretics: In IPAH, there is excessive afterload, resulting in RV dilatation and right heart failure. Diuretic therapy, often at high doses, may benefit patients with significant fluid overload.
- Arrhythmia management: Tachyarrhythmias are poorly tolerated and often manifest with worsening dyspnea, syncope, or right heart failure.15,16 The use of β-blockers is contraindicated in IPAH because it limits the ability to raise cardiac output by tachycardia.
In the last 10 to 15 years, a number of new disease-targeted therapies for PAH have been developed. These treatments are expensive and have significant adverse effects; however, they have been shown to improve symptoms and exercise capacity in IPAH, and some have been shown to improve survival.
The main classes of disease-targeted therapy for IPAH are as follows:
- Prostanoids: Prostanoids are analogs of prostacyclin and are available in intravenous (epoprostenol), nebulized (iloprost), and subcutaneous (treprostinil) forms. They are the treatment of choice for patients with severe (class IV) disease, and epoprostenol has been shown to improve survival in IPAH.17,18
- Phosphodiesterase-5 (PDE-5) inhibitors: PDE-5 inhibitors are oral agents that act by inhibiting the breakdown of cyclic guanosine monophosphate, thereby increasing the effect of locally produced nitric oxide. This results in inhibition of smooth muscle growth and pulmonary vasodilation. Sildenafil is the most widely used PDE-5 inhibitor at present.19
- Endothelin antagonists: Increased circulating levels of endothelin-1, a potent vasoconstrictor are observed in IPAH. Several oral agents are now available that can modify the endothelin system.20,21 Bosentan is a nonspecific endothelin antagonist, whereas ambrisentan and sitaxsentan are specific endothelin-A receptor blockers. Approximately 10% of patients on bosentan will have an elevation in hepatic transaminases, although the incidence is lower with the other endothelin antagonists.
It is now common practice to use combination therapy for patients with IPAH who have deteriorated despite targeted monotherapy. The rationale is that using multiple agents will be more effective in targeting the different pathophysiologic pathways that have been identified in IPAH. Approximately 30% of patients with IPAH are receiving combination therapy at present.
Atrial septostomy is thought to be of benefit in patients with IPAH who are in World Health Organization (WHO) functional class IV with right heart failure refractory to medical therapy.22 Atrial septostomy can decompress the RV, increase left ventricular preload, and increase cardiac output, particularly during exercise. This improves systemic oxygen transport despite the arterial oxygen desaturation.
Despite the development of disease-targeted therapy, there are still a significant number of patients who deteriorate on medical treatment, and these patients should be referred for transplantation.23 In the United Kingdom, current practice is for patients with IPAH requiring transplantation to receive bilateral lung transplantations. Recent changes in surgical technique, perioperative care, and immunosuppression have significantly improved outcome following transplantation, with survival rates of 86% at 1 year, 75% at 5 years, and 66% at 10 years now described in patients who undergo transplantation for IPAH.24
Imaging modalities, particularly echocardiography and cardiac magnetic resonance imaging (MRI), are becoming increasingly important in the follow-up of patients with IPAH, both in the setting of clinical trials and everyday clinical practice. Physicians have previously been reliant on WHO functional class, 6-minute walk test, biologic markers (such as N-terminal prohormone brain natriuretic peptide), and right heart catheterization to monitor response to treatment, all of which have acknowledged limitations.
Echocardiography is an attractive follow-up modality given that it is widely available, inexpensive, and safe. However, it is relatively operator dependent, and images may be suboptimal in patients with tachycardia, obesity, or poor acoustic windows. Cardiac MRI is now gaining widespread acceptance as the ideal marker in IPAH, providing abundant information regarding RV morphology and function in a safe, reproducible manner (Fig. 24–10). Extensive investigations are now under way to determine whether changes in CMR-derived RV functional parameters correspond to patient outcome.
Figure 24–10.
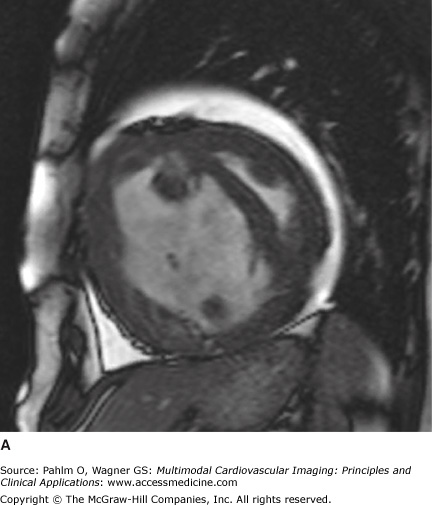
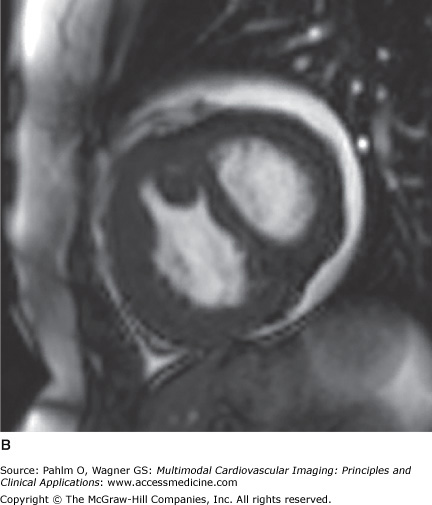
Short axis views of a patient with idiopathic pulmonary arterial hypertension at the time of diagnosis (A) and after 12 months of epoprostenol and sildenafil (B). Note the reduction in right ventricular dilatation, improved left ventricular filling, and resolution of the interventricular septal bowing.
A 33-year-old woman presented with an 18-month history of progressively worsening exertional dyspnea.
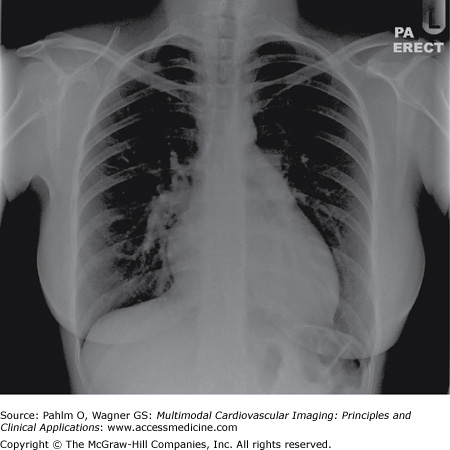
Clinical examination revealed a loud second heart sound and a tricuspid regurgitation murmur. ECG demonstrated right axis deviation and RV hypertrophy. Chest radiograph revealed cardiomegaly, prominent pulmonary arteries, and peripheral oligemia (Fig. 24–11).
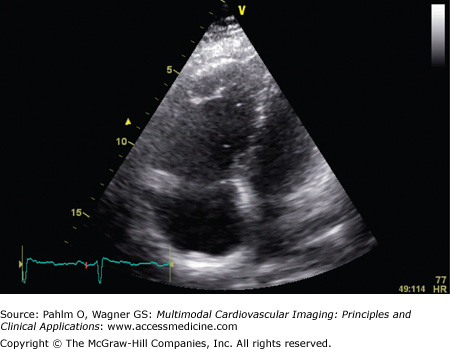
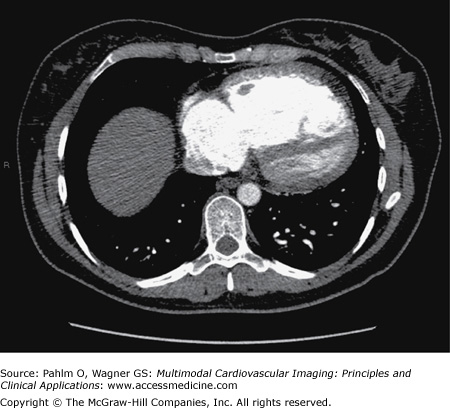
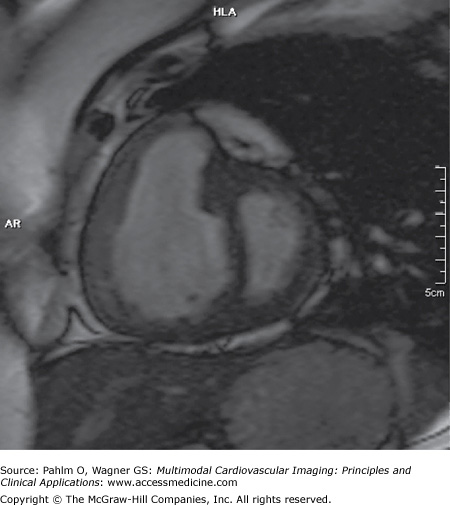
Her echocardiogram showed marked dilatation of the right-sided cardiac chambers and tricuspid regurgitation (Fig. 24–12). Her estimated PASP was 85 mm Hg. CT pulmonary angiogram revealed no evidence of pulmonary thromboembolic disease, but there was an enlarged main pulmonary artery and a dilated right heart (Fig. 24–13). Cardiac MRI demonstrated interventricular septal bowing and RV dilation, and planimetric evaluation revealed a markedly increased RVEDV, low LVEDV, and low stroke volume (Fig. 24–14).
She proceeded to right heart catheterization, where her mean PAP was elevated at 50 mm Hg, PVR was elevated at 12 Wood units, and cardiac output was low at 3.0 L/min. No other cause of PAH could be identified, so she was diagnosed as having IPAH. The patient was commenced on anticoagulation and the endothelin antagonist bosentan.
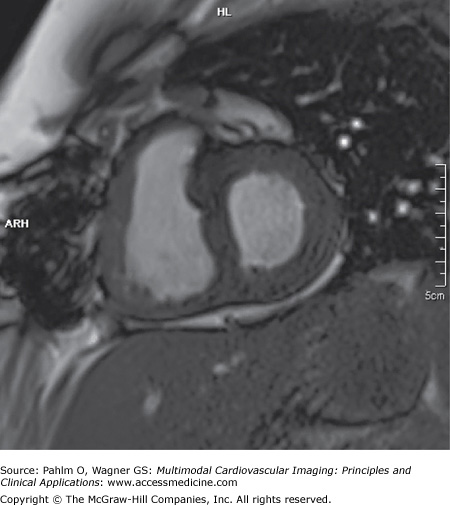
After 12 months of disease-targeted therapy, she had noticed a reduction in dyspnea, her 6-minute walk test had improved, and repeat cardiac MRI demonstrated an improvement in stroke volume and cardiac output, with a reduction in RV dilatation (Fig. 24–15).
Figure 24–15.
Related posts:
 Optical Mapping of Electrical Activity
Optical Mapping of Electrical Activity
 Phonocardiography
Phonocardiography
 Ischemic Heart Disease
Ischemic Heart Disease
 Development of the Heart, with Particular Reference to the Cardiac Conduction Tissues
Development of the Heart, with Particular Reference to the Cardiac Conduction Tissues
 Myocardial Perfusion Single Photon Emission Computed Tomography and Positron Emission Tomography
Myocardial Perfusion Single Photon Emission Computed Tomography and Positron Emission Tomography
 Cardiac Magnetic Resonance Imaging in Ischemic Heart Disease
Cardiac Magnetic Resonance Imaging in Ischemic Heart Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


