Mouse
fmol/mg protein a
[ 11 C]TBZ % inj dose/g b
Striatum
1,345
4.96
Thalamus
111
2.06
Hippocampus
141
2.01
Hypothalamus
411
2.96
Pons-medulla
167
1.96
Cortex (frontal)
115
1.91
Cerebellum
56
1.76
Rat
fmol/mg protein c
[ 11 C]DTBZ DV total d
Striatum
1,199
7.35
Thalamus
111–669
2.80
Hippocampus
124
2.51
Hypothalamic nuclei
838
5.13
Pons-medulla
–
–
Cortex (frontal)
103
2.18
Cerebellum
60
2.07
Human
fmol/mg protein a
[ 11 C]DTBZ DV total e
Caudate
742
12.5
Putamen
766
11.7
Hypothalamus
245
–
Hippocampus
83
–
Thalamus
–
4.75
Cortex (frontal)
57
4.16
Cerebellum
–
4.06
27.2.3 VMAT2 Substrates and Inhibitors
The VMAT2, being a transporter of multiple neurotransmitters (DA, NE, 5HT, histamine), has a fairly unspecific substrate specificity that extends to many biogenic amines, drugs of abuse (e.g., amphetamines), and neurotoxins (e.g., 1-methyl-4-phenylpyridinium, MPP+). The in vitro affinities of the transported molecules (endogenous neurotransmitters and amphetamines) for the [3H]DTBZ binding site are however quite low (>100 μmol). Compounds known to be antagonists for the receptors and neuronal membrane transporters for the monoamines are ineffective inhibitors of either VMAT2 transport function or high-affinity radioligand binding to the VMAT2 (Gonzalez et al. 1994; Wimalasena 2010).
When development of in vivo VMAT2 radioligands was begun, the list of known high-affinity ligands (agonist or antagonist) for the VMAT2 was limited to reserpine, tetrabenazine, and ketanserin. Reserpine, a complex alkaloid, has an extremely high affinity for VMAT2 (K D = 0.3 nM) but is essentially an irreversible inhibitor, with a protracted effect on brain VMAT2 (Naudon et al. 1995). Ketanserin has a moderate affinity for the VMAT2 (K D = 45 nM) but also high affinity for serotonin receptors, making it a less than optimal target for development of ligands with potential as in vivo imaging agents; attempts at modification of the ketanserin structure failed to yield higher affinity or more specific ligands (Darchen et al. 1988). Most recently, a series of compounds derived from lobelane and with moderate inhibitory activity towards the VMAT2 have been described (Zheng et al. 2005), but have yet been the target of radiotracer design.
Tetrabenazine (TBZ: 3-isobutyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-pyrido[2,1-a]isoquinolin-2-one, Fig. 27.1) is one of a series of compounds initially developed by Hoffman-LaRoche (Quinn et al. 1959); of a set of compounds with different alkyl substituents at the 3-position, it was selected for further development and eventually reached clinical use. For several decades following its clinical introduction, there had been little further examination of the structure-affinity relationships of these benzoisoquinolines for the VMAT2. It has only been more recently, and largely prompted by the interest in developing optimal radioligands for imaging, that a much larger set of derivatives of tetrabenazine have been synthesized and evaluated in vitro and in vivo. This includes DTBZ derivatives with 2-alkyl substituents (Lee et al. 1996), altered 3-alkyl groups (Zheng et al. 2011), and substitutions of longer alkyl or fluoroalkyl chains for the 9-O-methyl substituent (Goswami et al. 2006; Hostetler et al. 2007). Most importantly, however, and prompted entirely by the desire to optimize in vivo performance (see Sect. 27.4.3) was the resolution of dihydrotetrabenazine into active and inactive stereoisomers (Kilbourn et al. 1997), followed later by the synthesis and identification of all eight potential stereoisomers of DTBZ (Tridgett et al. 2012; Yao et al. 2011). Fortuitously, the isomer selected for the current generation of VMAT2 imaging agents turned out to be that with the highest affinity (K D = 1 nM) for VMAT2.
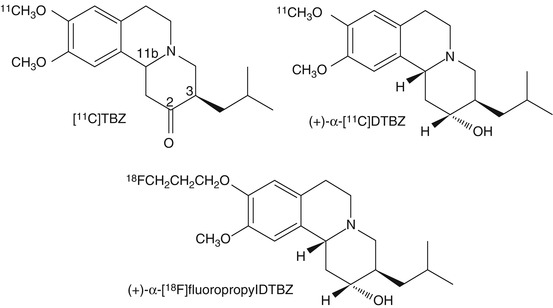
Fig. 27.1
Structures of tetrabenazine and PET radiopharmaceuticals currently in use in human PET studies of the VMAT2 in the human brain
27.3 VMAT2 Radioligands for Autoradiography
27.3.1 [3H]Tetrabenazine ([3H]TBZ)
Tritiated tetrabenazine was only recently reported by Rhee et al. (2011), using the catalytic hydrogenation of a suitably constructed alkene precursor (1,3,4,6,7,11b-hexahydro-3-(2-methyl-2-propenyl)-9,10-dimethoxy-2-oxo-2H-benzo[a]quinolizine) that upon reduction with tritium gas gave the doubly tritiated tetrabenazine with a specific activity of 1,850 GBq/mmol.
27.3.2 α-[3H]Dihydrotetrabenazine ([3H]DTBZ)
Most of the in vitro work using tritiated VMAT2 ligands has been performed with tritiated forms of α-dihydrotetrabenazine (termed TBZOH or DTBZ: 9,10-dimethoxy-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydrobenzo[a]quinolizin-2-ol, Fig. 27.1). This radioligand was first reported in 1981 (Scherman et al. 1981) and was prepared by the sodium borotritide reduction of the 2-ketone group of racemic tetrabenazine. The radioligand was thus obtained as a mixture of the α- and β-isomers resulting from a non-stereospecific reduction of the ketone. Although not clearly identified at the time, chromatographic isolation of a single isomer likely gave the α-(+/−)-[3H]dihydrotetrabenazine, in a specific activity of 185–555 GBq/mmol. More recently, the synthesis of the single isomer (+)-α-[3H]DTBZ was accomplished by O-methylation of (+)-α-9-O-desmethylDTBZ using [3H]methyl iodide, with a specific activity of 2,923 GBq/mmol (Kilbourn et al. 1997).
27.3.3 (+/−)-[3H]Methoxytetrabenazine ([3H]MTBZ)
In an attempt to prepare a radioligand with better radiochemical stability, the derivative 2-O-methyl-dihydrotetrabenazine was prepared in tritiated form (Vander Borght et al. 1995c). Alkylation of (+/−)-α-dihydrotetrabenazine with tritiated methyl iodide yielded the radioligand termed (+/−)-α-[3H]methoxytetrabenazine ([3H]MTBZ) in a specific activity of 3,034 GBq/mmol. This tritiated molecule was considerably more stable and showed less radiolytic decomposition upon storage as compared to tritiated dihydrotetrabenazine.
27.3.4 [3H]/[125I]Reserpine
Tritiated reserpine (292 Gbq/mmol) has been synthesized from [2,6-3H]3,4,5-trimethoxybenzoic acid and used for ex vivo autoradiography in rat brain. A radioiodinated reserpine derivative, 18-O-[3-(3-[125I]iodo-4-azidophenyl)-propionyl]methyl reserpate, has been used for photoaffinity labeling experiments with the VMAT2 (Sievert et al. 2007).
27.3.5 [125I]Iodovinyltetrabenazine
A radioiodinated derivative of tetrabenazine suitable for in vitro autoradiography was reported by Kung and coworkers (Kung et al. 1994). This was a dihydrotetrabenazine derivative with an iodovinyl substituent at the 2-position. The radioligand was prepared in very high specific activities (81,400 GBq/mmol) by an iodo-destannylation reaction and obtained as a mixture of two isomers, only one of which showed high-affinity binding to the VMAT2 (K d = 0.22 nM). The absolute stereochemistry of the radioiodinated ligand was not determined at that time, but based on the knowledge that has since been gained through the synthesis and characterization of all eight of the isomers of dihydrotetrabenazine, it is likely the higher-affinity compound had the 3R,11bR configuration but with the absolute configuration at the 2-position more difficult to predict as both 2R and 2S isomers of 2-substituted DTBZ derivatives have good binding affinities (Lee et al. 1996).
27.3.6 Ketanserin Derivatives
Although ketanserin is a nonspecific ligand for the VMAT2, it does have a high affinity for the transporter, and with appropriate pharmacological blocking of its other binding sites (e.g., serotonin receptors), two radioiodinated derivatives, 7-amino-8-[125I]iodoketanserin and 7-azido-8-[125I]iodoketanserin, have been used for in vitro autoradiography and photoaffinity labeling experiments (Darchen et al. 1989).
27.4 VMAT2 Radioligands for Positron Emission Tomography (PET) Imaging Studies
The development of VMAT2 radioligands for positron emission tomography (PET) began with tetrabenazine as the initial target compound. The choice was mostly practical. The molecule has two phenolic methoxy groups quite suitable for radiolabeling using carbon-11 methyl iodide, and the resultant radioligand would be unchanged from a well-known and characterized drug that had been in human use for decades; this averted any worries about toxicology, and the pharmacology of the drug was very well known. Interestingly, as will become evident in the following sections, the quest for optimal in vivo radioligand structures derived from tetrabenazine led to the synthesis of quite a number of derivatives, providing insights into the structure-activity relationships of the benzoisoquinolines and the identification of the absolute structures of the stereoisomers, which had never been done for this clinical drug.
27.4.1 [11C]Tetrabenazine ([11C]TBZ)
The labeling of tetrabenazine simply required removal of one of the O-methyl groups and replacement with the carbon-11 (t 1/2 = 20 min)-labeled methyl group (DaSilva and Kilbourn 1992; DaSilva et al. 1993b). Although simple in concept, the cleavage of the phenolic methoxy group using reagents such as boron tribromide proved to be a difficult reaction and led to complex mixtures of the mono- and bisdemethylated products. Careful chromatography yielded a single pure product assigned as the 9-O-desmethyltetrabenazine, with the assignment (9 vs. 10) based on proton and carbon-13 NMR spectroscopy; that assignment was later proved correct through a synthesis of the 9-O-desmethyltetrabenazine by an independent and unambiguous route. The reaction of the free phenolic compound with [11C]methyl iodide under basic conditions then yielded the desired [11C]tetrabenazine, in modest yields and specific activities (74,000–92,500 GBq/mmol) (DaSilva et al. 1993b). At the time of preparation, this radioligand was obtained as an unknown and uncharacterized mixture of stereoisomers; based on the data obtained in more recent studies (see below), the starting material and the final radiochemical product were both likely racemic mixtures of 3R,11bR and 3S,11bS-tetrabenazine.
27.4.2 (+/−)-α-[11C]Dihydrotetrabenazine ([11C]DTBZ)
[11C]Tetrabenazine was used for initial VMAT2 imaging studies, but consideration of its metabolic fate in the human body led to the rapid realization that what was really desired was the molecule termed dihydrotetrabenazine; this is the product of the first step in the metabolism of TBZ and is the predominant chemical species in the bloodstream after administration of the drug to rats or humans (Mehvar et al. 1987). Interestingly, this metabolite (termed TBZOH or DTBZ) had been used for years for in vitro studies of the VMAT2 (Scherman et al. 1988). The reduction of the ketone of TBZ by in vivo metabolism, or by chemical means in the laboratory, produces two isomers termed alpha- and beta-DTBZ; the desired higher-affinity isomer was the alpha stereoisomer but with absolute stereochemistry unknown. The required precursor for radiolabeling was now the 9-desmethyl-2-hydroxy derivative with the hydroxyl in the alpha configuration, prepared in two steps from TBZ by reduction of the ketone followed by demethylation (Jewett et al. 1997). Alkylation with [11C]methyl iodide produced (+/−)-α-[11C]DTBZ in moderate yields and useful specific activities (>59,200 GBq/mmol) but still as an undefined and likely racemic mixture of stereoisomers.
27.4.3 (+)-α-[11C]Dihydrotetrabenazine ((+)-α-[11C]DTBZ)
As the last step in the optimization of a specific VMAT2 radioligand, the stereoisomers of α-DTBZ were separated using preparative chiral HPLC (Kilbourn et al. 1995b, 1997). There were found to be only two isomers which rotated polarized light in opposite directions, assigned as the (+)- and (−)-isomers. Crystals of the (−)-DTBZ isomer were successfully grown for X-ray structural analysis, and the absolute stereochemistry of these isomers finally assigned: (2R,3R,11bR) for the (+)-isomer and (2S,3S,11bS) for the (−)-isomer (Kilbourn et al. 1997). In vitro binding assays showed that these two isomers had drastically different binding affinities, with only the (+)-isomer showing high-affinity binding (1 nM) and a very low affinity (4,000 nM) for the (−)-isomer. More recently, efforts have been successful at the synthesis and identification of all eight of the possible stereoisomers of dihydrotetrabenazine (Tridgett et al. 2012; Yao et al. 2011). Subsequent syntheses of all of the isomers and determination of the in vitro binding affinities have shown that the isomer with the 2R,3R,11bR absolute stereochemistry is the highest-affinity DTBZ ligand for the VMAT2 (Yao et al. 2010).
The synthesis of (+)-α-[11C]DTBZ then required the preparation of the single resolved isomer of the 9-O-desmethylDTBZ, which was first accomplished by a low-yield sequence of reduction of TBZ, chiral column resolution of DTBZ isomers, and a modified demethylation reaction condition (sodium hydride/N-methylaniline/hexamethylphosphoramide) (Kilbourn et al. 1997). More recently, improved synthetic routes to TBZ, (+)-α DTBZ, and (+)-α-9-desmethylDTBZ have been published (Boldt et al. 2008; Paek et al. 2010; Rishel et al. 2009a; Yu et al. 2010), making the synthesis of the precursor to the desired radioligand much more practical. Alkylation of the desmethyl precursor with [11C]methyl iodide yields the currently routinely used radioligand, (+)-α [11C]DTBZ, in high average specific activities (370,000 GBq/mmol) and a defined 2R,3R,11bR absolute stereochemistry.
27.4.4 [11C]Methoxytetrabenazine ([11C]MTBZ)
In an attempt to alter and perhaps improve the metabolic disposition of VMAT2 radioligands, a new derivative with a [11C]methoxy substituent at the 2-position was synthesized (DaSilva et al. 1993c). Prepared by reaction of (+/−)-DTBZ with [11C]methyl iodide under strong basic conditions, the radioligand 2-O-methoxydihydrotetrabenazine (MTBZ) was successfully introduced into human studies (Vander Borght et al. 1995b). However, the radiochemical synthesis proved more difficult than the alkylation of the phenol (as for DTBZ), and with the development of (+)-α [11C]DTBZ, MTBZ was not extensively studied although it is excellent as an in vitro ligand when prepared in tritiated form.
27.4.5 Fluorine-18-Labeled Dihydrotetrabenazines
The carbon-11-labeled ligands and in particular (+)-α-[11C]DTBZ have now been used in a wide variety of clinical studies of neurological and psychiatric diseases. The application of VMAT2 imaging to clinical care, however, was likely to require the availability of radioligands labeled with longer half-life radionuclides. Fortunately, development of fluorine-18 (t 1/2 = 109.6 min)-labeled VMAT2 radioligands was able to build on and extend the chemistry and pharmacology developed in the carbon-11 series.
The substitution of the 9-methoxy group of DTBZ with [18F]fluoroalkoxy groups resulted in the successful synthesis of 9-O-[18F]fluoromethyl-, 9-O-(2-[18F]fluoroethyl)- and 9-O-(3-[18F]fluoropropyl)DTBZ (Goswami et al. 2006; Hostetler et al. 2007; Kung et al. 2007). The [18F]fluoromethyl derivative (K i = 0.87 nM for rat VMAT2) was prepared by O-alkylation of (+)-α-9-O-desmethylDTBZ with [18F]FCH2Br under basic conditions (Hostetler et al. 2007). The [18F]fluoroethyl (K i = 0.76 nM) and [18F]fluoropropyl (K i = 0.56 nM) derivatives were first prepared in racemic form by [18F]fluoride ion displacement of the corresponding mesylates (Fig. 27.1) in specific activities >74,000 GBq/mmol. Subsequently, a stereospecific synthesis of (+)-α-9-O-(3-[18F]fluoropropyl)DTBZ (also termed [18F]AV-133: (2R,3R,11bR)-9-(3-[18F]fluoropropoxy)-10-methoxy-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo[a]quinolizin-2-ol) from the corresponding chirally resolved mesylate was developed, yielding the very high-affinity (K i = 0.1 nM) single isomer, with specific activities of 55,000–74,000 GBq/mmol. As was expected, the (−)-α-(3-fluoropropyl)DTBZ isomer showed negligible affinity for VMAT2 (>3,000 nM). Recently, in an attempt to improve on the in vivo defluorination of [18F]fluoroethylDTBZ, the radioligand was synthesized using tetradeuterated [18F]fluoroethyl bromide; the deuterated ligand showed regional brain pharmacokinetics similar to [11C]DTBZ with slightly higher total uptake and higher striatal binding potentials (Varone et al. 2012).
In the pursuit of improved properties for in vivo imaging, Kung and coworkers have explored several other derivatives of TBZ derivatives. Replacement of the 2-hydroxyl group of DTBZ with a 2-fluoropropylamino group yielded a derivative with an in vitro affinity of 7 nM (as racemate) (Zhu et al. 2009). Substitution of the 2-hydroxyl function of 2-[18F]fluoropropyl-(+)-DTBZ with an epoxide ring provided a derivative with an excellent in vitro affinity (0.08 ± 0.01 nM) (Kung et al. 2008).
A completely different approach to fluorine-18-labeled DTBZ molecules was reported in a series of patents (Amarasinghe et al. 2009; Kankanamalage et al. 2009; Rishel et al. 2009b). A very large number of fluoro-substituted (+)-α-DTBZ derivatives are claimed in this series of patents, but in vitro biological information is only available for a few of the compounds claimed. The 2-fluoroethyl-substituted DTBZ derivative shows reasonable affinity (K i = 19 nM), consistent with the earlier demonstration of bulk tolerance of reasonably small 2-alkyl substituents (Lee et al. 1996). Similarly, the 2-fluoroethoxy ether derivative has a good in vitro binding affinity of 3.2 nM, not significantly different than that previously obtained for the slightly smaller methyl ether derivative (3.9 nM; Vander Borght et al. 1995c).
Of the numerous fluorine-18 radiotracers that have been prepared, only the 9-(3-[18F]fluoropropyl)-dihydrotetrabenazine has been reported to be used in human imaging studies.
27.5 Evaluation of VMAT2 Imaging Radioligands in Animals
The evaluation of VMAT2 radioligands for pharmacological specificity, kinetics, sensitivity to endogenous neurotransmitters, propensity for up- or downregulation, and changes in models of human disease has been extensively pursued in both small and large animal models.
27.5.1 Mouse Brain VMAT2 Studies
The mouse was utilized for the initial evaluation of [11C]TBZ, where it was demonstrated that the (a) radioligand localized in regions of the brain containing high concentrations of the VMAT2 (b) localization was likely not specific only to dopaminergic neurons but, as expected, also due to noradrenergic and serotonergic innervation (Table 27.1), and (c) binding could be blocked by pharmacological doses of TBZ but not drugs that bound to the neuronal membrane dopamine transporter (DaSilva and Kilbourn 1992). These simple initial studies also demonstrated the reversibility of [11C]TBZ binding in the rodent brain.
Subsequent studies of VMAT2 radioligand binding in the mouse brain demonstrated that both simple measurement of tissue ratios between high (striatum) and low (cerebellum) regions of the mouse brain, and more quantitative methods using PET imaging, could be used to show decreases of VMAT2 binding sites after MPTP (1-methyl-4-phenyl-1,2,3,4-terahydropyridine) lesioning (Chao et al. 2012; Kilbourn et al. 2000). The utility of in vivo radioligand binding methods to demonstrate increases of VMAT2 binding was accomplished through studies of genetic mutants, known from in vitro studies to overexpress the VMAT2 in the brain (Kilbourn et al. 1995a).
27.5.2 Rat Brain VMAT2 Studies
A larger number of studies of in vivo VMAT2 radioligands have been conducted in rats. These have utilized a variety of techniques, including (a) ex vivo measurements of tissue distributions following bolus injections, (b) ex vivo autoradiography of rat brain slices (Tsao et al. 2010), (c) ex vivo determination of regional brain distributions after equilibrium infusions (Kilbourn and Sherman 1997), and (d) in vivo PET imaging studies including dynamic multi-tracer approaches (Kilbourn et al. 2007; Sossi et al. 2009), in attempts to characterize the distribution, pharmacology, and sensitivity of the VMAT2 radioligand methods.
As with the studies in mice, determinations of regional brain distributions of VMAT2 radioligands in rats show excellent correlations with the published in vitro distribution of VMAT2 binding sites (Table 27.1). Use of both the active and inactive isomers of [11C]DTBZ has shown that uptake and retention of VMAT2 radioligands in regions of the rat brain such as the cerebellum and the occipital cortex represent essentially negligible concentrations of the VMAT2 (Kilbourn and Sherman 1997) and thus are reasonable regions for estimations of nonspecific distributions of these radiotracers.
The sensitivity of measures of VMAT2 radioligand binding to regulation by chronic drug use has been examined using both in vitro and in vivo rat studies, with decidedly mixed results. Numerous studies have demonstrated that chronic administration of such drugs as L-DOPA/benserazide, apomorphine, tetrabenazine, pramipexole, paroxetine, amphetamine, or cocaine did not alter in vitro levels of VMAT2 radioligand binding, nor was in vivo DTBZ binding altered by repeated administration of L-DOPA, deprenyl, or tetrabenazine (Kemmerer et al. 2003; Kilbourn et al. 1996; Schwartz et al. 2006; Vander Borght et al. 1995a; Vilpoux et al. 2000; Wilson and Kish 1996). These results supported the hypothesis that VMAT2 was a less regulated site than, for example, the neuronal membrane dopamine transporter, for which changes could be seen following such drug treatments (Vander Borght et al. 1995a). In contrast, several other studies have reported increases or decreases of VMAT2 radioligand binding after administration of some of the same or related drug substances (Frey et al. 1997; Schwartz et al. 2006; Sossi et al. 2010): for methamphetamine, reductions of VMAT2 binding are observed after neurotoxic but not lower behaviorally effective doses (Frey et al. 1997). A few studies in rats have supported a sensitivity of VMAT2 radioligand binding in vivo to alterations of endogenous dopamine levels following administration of drugs which directly decrease endogenous dopamine levels (α-methyl-p-tyrosine, AMPT) (Kilbourn et al. 2008, 2010; Tong et al. 2008) or indirectly raise dopamine levels (pramipexole) (Sossi et al. 2010). Those studies have indicated that alterations of vesicular dopamine concentrations may in fact result in changes of in vivo VMAT2 radioligand binding, and the increases of VMAT2 radioligand binding can be reversed by replenishment of vesicular dopamine levels using exogenous L-DOPA (Kilbourn et al. 2010). It should be noted that modest changes of in vivo radioligand binding (36 % increase) require significant depletion of endogenous dopamine concentrations (>75 %) in the rat brain.
27.5.3 VMAT2 Radioligand Studies in Nonhuman Primates
In the normal monkey brain, VMAT2 radioligands show high in vivo binding in the striatal regions, with lower levels of radioactivity retention in the thalamus, hypothalamus, and midbrain cell bodies (raphe nucleus, locus ceruleus, substantia nigra) (DaSilva et al. 1993a; Kilbourn et al. 2007) and negligible in cortex and cerebellum. The binding is stereospecific with low and uniform brain distribution of the corresponding inactive stereoisomers (Kilbourn et al, unpublished data). A comprehensive study of the in vitro distribution of VMAT2 radioligand binding site in the nonhuman primate brain is still missing from the literature, but specific binding in the caudate and putamen has been demonstrated using in vitro autoradiography (Chen et al. 2008; Tian et al. 2012), and the in vivo distribution of binding would be consistent with the expected distribution of the dopaminergic, noradrenergic, and serotonergic systems as seen in rodents and human brain (Table 27.1).
An early study of [11C]tetrabenazine in a monkey with a unilateral MPTP-injected monkey demonstrated the complete loss of in vivo radioligand binding in that severe example of dopaminergic neuronal degeneration (DaSilva et al. 1993a). Subsequent studies have used improved imaging instrumentation, the better radioligand (+)-α-[11C]DTBZ, large numbers of monkeys, and multi-tracer studies to better characterize the MPTP-induced loss of in vivo VMAT2 binding sites in this animal model of Parkinson’s disease. Using a multi-tracer approach ((+)-[11C]DTBZ, [11C]methylphenidate (for the neuronal membrane dopamine transporter, DAT) and [18F]fluoroDOPA), Doudet and coworkers (2006) demonstrated an age-related decrease of VMAT2 radioligand binding in a small cohort of monkeys and that the measures of losses of DTBZ binding were the most sensitive of the three radiotracer methods for detecting striatal changes in asymptomatic, mild unilateral MPTP intoxication. Similar results were obtained in a study using [11C]DTBZ (for VMAT2) and [11C]WIN 35,428 (for DAT) in asymptomatic MPTP-treated monkeys, where significant (50 %) losses of VMAT2 radioligand binding preceded changes in the DAT (Chen et al. 2008). In a monkey model of progressive Parkinsonism induced by repeated MPTP administration spread over several months, losses of DTBZ binding to the VMAT2 were detectable earlier at the less affected stages and then also in monkeys that showed behavioral recovery, than with fluoroDOPA (Blesa et al. 2010). Consistently, unilateral administration of MPTP results in significant decreases in [11C]DTBZ binding (as well as other dopaminergic markers such as DAT radioligands and 6-[18F]fluoroDOPA) and the extent of the decreases agree with postmortem autoradiographic studies of monkey brains after MPTP lesioning (Tian et al. 2012).
In a single study, Fantegrossi et al. (2004) examined the potential changes of in vivo [11C]DTBZ binding in a primate model of self-administration of methylenedioxymethamphetamine (MDMA) and found no evidence of dopaminergic degeneration in the striatum. In contrast, a postmortem study of amphetamine-treated baboons showed evidence of neuronal toxicity in the form of reduced binding of [3H]DTBZ to the VMAT2 (and also reduced radioligand binding to the DAT) (Ricuarte et al. 2005).
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 Progress in PET Imaging of the Norepinephrine Transporter System
Progress in PET Imaging of the Norepinephrine Transporter System
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 PET and SPECT Imaging of Steroid Hormone Receptors
PET and SPECT Imaging of Steroid Hormone Receptors
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


