(1)
Experimental Therapeutics and Molecular Imaging Laboratory, Department of Neurology, Neuroscience Center, Massachusetts General Hospital, Boston, MA, USA
(2)
Program in Neuroscience, Harvard Medical School, Boston, MA, USA
(3)
Renal Division, Transplantation Research Center, Brigham and Women’s Hospital and Children’s Hospital, Harvard Medical School, Boston, MA, USA
Abstract
Regulatory T cells (Tregs) are amongst the most widely studied cells in a variety of immune-mediated conditions, including transplantation and Graft Versus Host Disease (GVHD), cancer and autoimmunity; indeed, there is great interest in the tolerogenic potential of Treg-based therapy. Consequently, the need to establish the mechanisms that determine Treg survival and longevity, in addition to developing new tools to monitor these parameters, is paramount. Using both a mouse model of GVHD and a mouse model of Type 1 Diabetes (T1D), we describe herein a dual reporter system based on Gluc and multiplexed with SEAP and non-secreted Firefly luciferase (Fluc), which permits simultaneous imaging and noninvasive tracking of two different T-cell populations (CD4+CD25+ Tregs and CD4+CD25− Tcon cells) in vivo by transducing the cells with different lentiviruses bearing distinct color signatures. This new technology promises to overcome the limitations of the conventional methods currently available to study lymphocyte survival in vivo. Furthermore, this novel technique has applications not only in autoimmunity and alloimmunity, but also in the wider field of immunology.
Key words
LuciferaseBioluminescence imagingLymphocytesTregsHomeostasisSurvivalGVHDType 1 diabetesGrant K. Lewandrowski and Ciara N. Magee contributed equally to this work.
1 Introduction
The discovery of CD4+CD25+ regulatory T cells (Tregs) in 1995 was accompanied by compelling evidence of their critical role in the maintenance of self-tolerance and prevention of autoimmunity [1]. These cells, characterized by the expression of the transcription factor FoxP3, comprise approximately 5–10 % of CD4+ T cells in normal naive mice and are of thymic origin (natural Tregs; nTregs) [1, 2]. Studies have shown that it is possible to induce Tregs from naïve peripheral CD4+CD25− T cells, depending on the stimulus and cytokine milieu [3], and that these adaptive Tregs (iTregs) also possess the ability to regulate and suppress the immune response, even in the absence of nTregs [4]. Treg therapy has emerged as a promising tolerogenic approach for a variety of immune-mediated conditions, including autoimmune disease and Graft-Versus-Host-Disease (GVHD); in transplantation models, Tregs have been shown to actively regulate allogeneic T effector (Teff) cell responses and promote transplantation tolerance [5, 6]. Although our knowledge of the wide spectrum of their suppressive properties is constantly expanding [7, 8], the homeostatic mechanisms involved in maintaining the population and functions of Tregs have been largely unexplored [9]. Given the increasing number of clinical trials testing the safety and efficacy of Treg-based immunotherapy [10], the need to establish the mechanisms that determine the survival and longevity of Tregs, and, consequently, the means of monitoring these parameters, is paramount.
Secreted reporters in the blood or serum have been shown to be important tools for the detection, quantitation and noninvasive monitoring of different biological processes, including cell tracking, in experimental models [11–18]; indeed, they have several advantages over conventional in vivo imaging methods for monitoring gene expression [19, 20]. In culture, secreted reporters can be quantified in cell-free conditioned medium without the need for cell lysis, thereby leaving the cells intact and available for confirmation analysis. In vivo, secreted reporters can be easily monitored in animals by withdrawing a few microliters of blood or urine and assaying for their activity, allowing real-time monitoring of in vivo processes; furthermore, bioluminescence imaging can be performed to localize cell activity in vivo, although this method requires expensive instrumentation (cooled CCD camera), is time-consuming, involves frequent anesthesia and repeated systemic substrate injections, and is limited by photon absorption by tissues as a function of depth [20].
The most commonly used serum reporter is the secreted alkaline phosphatase or SEAP [12, 13, 15–17, 21–25]. Normally, alkaline phosphatase is not secreted; however, it has been shown that placental alkaline phosphatase is expressed and released efficiently into the conditioned medium of mammalian cells that have been engineered to express this reporter [21]. The level of SEAP in the conditioned medium is linearly related to the changes in its intracellular mRNA level as well as cell number [21, 22]. It is also possible to quantitatively detect SEAP in the serum of animals. In 1997, Abruzzese et al. used the SEAP serum level to monitor the induction of gene expression followed by gene transfer to the muscles of adult mice [26]. Since then, the SEAP reporter assay has been used as a surrogate serum marker to monitor different in vivo processes including tumor growth and drug efficacy [13], DNA vaccination [27]; promoter and transcription factors activation and inhibition [12, 16, 25]; viral gene transfer [28]; inflammatory events [17]; viral infection and replication [29]; and endoplasmic reticulum stress [23].
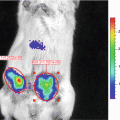
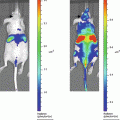
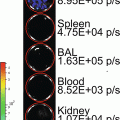
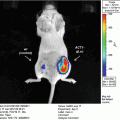
We have recently established and reported a novel secreted luciferase reporter from the marine copepod Gaussia princeps known as Gaussia luciferase (Gluc) [11, 14, 18, 30, 31]. We have shown that this reporter is naturally secreted from mammalian cells in an active form and that the levels of Gluc in the conditioned medium correlate linearly with respect to cell number, growth and proliferation [11, 31]. The Gluc reporter system has several advantages over the SEAP assay including a much reduced assay time (1–10 min vs. 1.5–2 h), greatly increased sensitivity (20,000-fold in vitro and 1,000-fold in vivo), and a linear range covering over 5 orders of magnitude of cell number. In vivo, the Gluc reporter assay has been used to localize tumors and to monitor tumor formation and proliferation using bioluminescence imaging [31, 32]. Since Gluc is naturally secreted, we have shown that its level in the blood or urine can be used as a marker to monitor different in vivo biological events such as tumor growth and response to therapy, viral infection and replication as well as the viability of circulating cells [18]. This is a crucial advantage when investigating the behavior of widely circulating cells where a lack of focal accumulation/cell density precludes the use of typical imaging techniques to localize the cells; in these instances, blood or serum levels can be used to reflect cell activity.
We describe herein a dual reporter system based on Gluc and multiplexed with SEAP and non-secreted Firefly luciferase (Fluc) which permits simultaneous imaging and noninvasive tracking of two different T-cell populations (CD4+CD25+ Tregs and CD4+CD25− Tcon cells) in vivo by transducing the cells with different lentiviruses bearing distinct color signatures. One further advantage offered by this method of cell identification is that even if the cell loses its normal identifier due to cell plasticity, it is still possible to identify the original lineage of the cell, as it will retain its transduced color signature. This has become particularly important as we become more aware of cell plasticity, e.g., induced Tregs arising from conventional T cell precursors can lose FoxP3 expression and regulatory function over time, becoming effector T cells [33].
2 Materials
2.1 Large-Scale Lentiviral Packaging and Concentration
1.
Dulbecco’s Modified Eagle Medium (DMEM): High glucose, HEPES, 10 % Fetal Bovine Serum, 1 % Penicillin/Streptomycin. Store at 4 °C.
2.
Opti-MEM® Reduced Serum Medium: GlutaMAX, 1 % Penicillin/Streptomycin. Store at 4 °C.
3.
0.05 % Trypsin–EDTA. Store at 4 °C.
4.
Sterile Phosphate Buffered Saline (PBS).
5.
2 M Calcium Chloride solution.
6.
2× HEPES Buffered Saline solution (HeBS): 0.28 M NaCl, 0.05 M HEPES, 1.5 mM Na2HPO4 anhydrous, distilled H2O up to 1 L, pH 7.00 (see Note 1).
7.
2.5 mM HEPES solution.
8.
pCMV-ΔR8.91 plasmid (Addgene, Cambridge, MA, USA).
9.
pCMV-VSV-G plasmid (Addgene, Cambridge, MA, USA).
13.
Standard treated 15 cm cell culture dishes.
14.
SW32 Rotor Tubes: Thinwall, Polyallomer, 38.5 mL, 25 × 89 mm.
15.
Standard conical tubes (50 mL).
16.
PVDF Vacuum Filtration Assembly: High volume, 0.45 μm.
17.
Cultured HEK/293T (containing SV40 Large T-antigen) cell line.
18.
Standard vortex mixer.
19.
Coulter counter or hemacytometer.
20.
Trypan blue.
21.
Tabletop centrifuge.
22.
Microcentrifuge.
23.
Ultracentrifuge.
24.
SW32 swinging bucket rotor.
25.
Confocal Laser Scanning Microscope.
2.2 T Cell Isolation and Activation
1.
MACS Buffer: 0.5 % Bovine Serum Albumin, 2 mM EDTA, Sterile PBS. Store at 4 °C.
2.
RPMI-1640 Medium: 10 % Fetal Bovine Serum, 1 % Penicillin/Streptomycin, 1 % l-Glutamine. Store at 4 °C.
3.
70 μm filter units.
4.
500 mL Rapid-Flow Filter Unit (Thermo Fisher, Waltham, MA, USA).
5.
Standard conical tubes (15 mL).
6.
Syringes (1 mL).
7.
ACK (Ammonium-Chloride-Potassium) lysis buffer (GIBCO, Grand Island, NY, USA).
8.
MACS LD & MS columns (Miltenyi Biotec, Cambridge, MA, USA).
9.
Mouse Regulatory T cell Isolation Kit (Miltenyi Biotec, Cambridge, MA, USA).
10.
Dynabeads: Mouse T-Activator CD3/CD28 beads (GIBCO, Grand Island, NY, USA).
2.3 Lentiviral Infection of T Cells
1.
Polybrene. Resuspend to 8 mg/mL stock. Store at 4 °C.
2.4 Bone Marrow Extraction
1.
Sterile Petri dishes.
2.
Dissection scissors.
3.
Forceps.
4.
Razor blades.
5.
Syringes (5 mL).
6.
Needles (27 Gauge).
7.
CD3ε MicroBead Kit (Miltenyi Biotec, Cambridge, MA, USA).
8.
MACS LD columns (Miltenyi Biotec, Cambridge, MA, USA).
2.5 Injection of Cells
1.
Magnetic bead separation rack (New England Biolabs, Beverly, MA, USA).
2.
U-100 insulin syringes (28.5 Gauge).
2.6 Bioluminescent and Chemiluminescent Assays and Bioluminescent Mouse Imaging
2.
Hydrochloric acid.
3.
Methanol.
4.
Opaque white 96-well assay plates.
5.
Great EscAPe SEAP Reporter System 3 (Clontech, Mountain View, CA, USA).
6.
7.
Nude athymic mice.
8.
Isoflurane mouse anesthetic.
9.
50 mM Ethylenediaminetetraacetic acid (EDTA).
10.
Microcentrifuge tubes.
11.
Luminometer.
12.
Cooled charge-coupled device (CCD) camera.
13.
XGI-8 gas anesthesia system.
14.
Living Image software.
3 Methods
3.1 Large-Scale Lentiviral Packaging and Concentration
3.1.1 Day 1
3.1.2 Day 2
1.
Aspirate out existing medium from all plates and replace with 20 mL of supplemented DMEM medium one hour prior to transfection.
2.
Prepare Calcium-Phosphate transfection mix in two separate 50 mL conical tubes. To Vial 1 add the following: 180 μg desired plasmid vector, 180 μg pCMV-ΔR8.91 plasmid, 120 μg pCMV-VSV-G plasmid, 969 μL 2M CaCl2 solution, and bring total volume up to 7,800 μL with 2.5 mM HEPES solution. To Vial 2 add 7,800 μL 2× HeBS solution.
3.
Mix contents of Vial 1 drop-wise to Vial 2 while vortexing at medium speed. Tightly cap Vial 2 without ceasing vortexing and increase vortex to max speed for 1 min (see Note 9).
4.
Incubate Vial 2 at room temperature for 20 min.
5.
After incubation, add 1.5 mL of transfection mixture drop-wise to each of ten plates of 293T cells and gently rock plates to evenly distribute the mixture.
3.1.3 Day 3
1.
Wash plates by aspirating off existing medium and replacing with 13 mL supplemented Opti-MEM medium (see Note 10) for each plate. Carefully pipette down sides of plate to avoid disrupting cell monolayer.
2.
Aspirate off the 13 mL of supplemented Opti-MEM and add an additional 13 mL of supplemented Opti-MEM to each plate.
3.
Return plates to incubator for 24 h.
3.1.4 Day 4
1.
Harvest the 13 mL of virus-saturated medium from each plate and store at 4 °C in 50 mL conical tubes. Refresh plates with additional 13 mL supplemented Opti-MEM and incubate for 24 h.
3.1.5 Day 5
1.
Harvest 13 mL virus-saturated medium from each plate, combine with harvested medium from Day 4, and centrifuge at 450 × g for 5 min.
2.
Decant supernatant and filter using a 0.45 μm vacuum filtration assembly. Transfer filtrate to SW32 ultracentrifuge tubes.
3.
Ultracentrifuge the filtrate at 133,907 × g (max) for an SW32Ti Rotor for 98 min at 4 °C.
4.
Aspirate supernatant and resuspend all viral pellets using a total of 200 μL supplemented Opti-MEM. Aliquot and store at −80 °C.
3.2 T Cell Preparation
3.2.1 Day 1: T Cell Isolation and Activation
3.
Harvest the spleens from the relevant mice using aseptic technique; store them temporarily in a 15 mL conical tube containing sterile PBS.
4.
Prepare a single-cell suspension by first placing the cell strainer in a 50 mL conical tube and adding the spleens into the strainer.
5.
Mechanically dissociate the spleens through the cell strainer using the flat edge of the syringe plunger. Rinse the cell strainer frequently with sterile PBS.
6.
Add sterile PBS to a total volume of 45 mL and centrifuge at 300 × g for 6 min.
7.
Discard the supernatant and add 1–2 mL of ACK lysis buffer per spleen, making sure to pipette thoroughly until the cell pellet is completely resuspended.