Etiology
Sjögren’s syndrome, or “sicca syndrome,” is a disorder of the immune system that is largely defined by its two most common symptoms—dry eyes and a dry mouth; these symptoms frequently accompany other autoimmune disorders. Distinction is usually made between Sjögren’s syndrome that occurs in the absence of an accompanying connective tissue disease (primary Sjögren’s syndrome) and Sjögren’s syndrome accompanied by another connective tissue disease (secondary Sjögren’s syndrome) such as systemic lupus erythematosus, rheumatoid arthritis (RA), scleroderma, systemic sclerosis, cryoglobulinemia, and polyarteritis nodosa. Both primary and secondary types occur with similar frequency, and virtually all organs may be involved.
The etiology of the disease is not fully understood, but it is speculated that environmental factors trigger the human leukocyte antigen (HLA)-DR–dependent immune system, which affects the exocrine glandular vascular endothelium. Infiltration of the exocrine glands by lymphocytes leads to apoptosis and glandular dysfunction. There is evidence that certain HLA class II antigens are increased in these patients; in Caucasian individuals, Sjögren syndrome is associated with HLA-DR3 and certain HLA-DQ alleles. Possible triggering environmental factors include viruses, such as Epstein-Barr, human T-lymphotropic virus-1, human herpesvirus 6, human immunodeficiency virus, hepatitis C, and cytomegalovirus. Damage or cell death caused by viral infection or other causes may provide triggering antigens to Toll-like receptors on dendritic or epithelial cells.
Prevalence and Epidemiology
Sjögren syndrome can develop at any age, but 59 years of age is the average at diagnosis (range, 43–75 years). There are two age peaks for primary disease—the first in the third decade and the second after menopause. The worldwide prevalence of Sjögren syndrome is 14.4 per 100,000 population, and the condition is nine times more frequent in women than in men.
Clinical Presentation
Keratoconjunctivitis, the characteristic ophthalmologic feature of Sjögren syndrome, is caused by a lack of tear film secondary to destruction of the lacrimal gland. Patients complain of dry eyes, irritation, or photophobia. Careful assessment of patients with dry eyes is important; in particular, the integrity of the corneal surface needs to be evaluated.
Dryness of the mouth makes swallowing and talking difficult. Sudden swelling of a salivary gland suggests infection, whereas a more insidious onset of salivary gland enlargement can signal lymphomatous transformation.
Exocrine glandular involvement is typically the first manifestation of Sjögren syndrome, with a subsequent slow deterioration in salivary and lacrimal function. In a cohort of patients with primary disease, musculoskeletal (60%), urogenital (40%), hematologic (24%), cutaneous (20%), pulmonary (11%), gastrointestinal (7%), neurologic (8%), and renal (3%) manifestations were noted. Symptoms of myalgia and arthralgia are frequent, whereas arthritis is rare. There are few descriptions of the type of arthritis found in these patients, but it tends to be similar to RA. Urogenital symptoms are secondary to vulval dryness and pruritus, which leads to dyspareunia. Many patients with primary Sjögren syndrome have a normocytic, normochromic anemia, and leukopenia, as well as an increased risk for hematologic malignancies (leukemia, myeloma, Hodgkin and non-Hodgkin lymphoma). Given that the syndrome is characterized by a lymphocytic infiltrate, it can be anticipated that a spectrum of lymphoproliferative disorders may develop in patients with Sjögren syndrome, including lymphocytic (lymphoid) interstitial pneumonia (LIP), follicular bronchiolitis, nodular lymphoid hyperplasia, and mucosa-associated lymphatic tissue (MALT) lymphoma, in addition to frank lymphoma. Amyloid deposition in conjunction with LIP has also been described.
The incidence of cutaneous vasculitis has been estimated at 9%, and it is usually manifested as purpura, urticarial lesions, and maculopapules. Raynaud phenomenon has been reported in 30% of patients, and in patients with primary Sjögren syndrome and Raynaud phenomenon, the density of nail fold capillaries is significantly reduced.
Patients with primary Sjögren syndrome have an increased risk for additional autoimmune diseases, particularly RA, scleroderma, and primary biliary cirrhosis. Clinical or biochemical evidence of liver disease, or both, is found in up to 10% of patients, and the incidence of Sjögren syndrome in patients with chronic liver disease suggests a link between hepatitis C virus and Sjögren syndrome.
Pathophysiology
Pathology
In one of the few computed tomography (CT)-pathology correlative studies of primary Sjögren syndrome, the major histologic patterns of lung disease found on biopsy were nonspecific interstitial pneumonia (NSIP) (61%), bronchiolitis, type not specified (12%), malignant lymphoma (12%), amyloidosis (6%), atelectatic fibrosis (6%), and honeycomb fibrosis, histologic subtype not specified (3%). The CT appearance of an NSIP pattern had a high positive predictive value for the presence of NSIP on biopsy. The relative scarcity of usual interstitial pneumonia (UIP) in this study may be a result of the small size of the series and also because those with typical UIP on high-resolution CT (HRCT) may not have undergone surgical lung biopsy. Conversely, in a smaller series of lung biopsy specimens ( n = 9), two-thirds of the patients had histologic UIP, and a third had NSIP.
Malignant lymphoma (see Chapter 24 ) is the major cause of morbidity and mortality in patients with primary Sjögren syndrome, and these patients have a 44-fold increased risk for the development of non-Hodgkin lymphoma.
LIP is a diffuse interstitial proliferation of mature small lymphocytes and plasma cells, and it commonly occurs in patients with Sjögren syndrome, autoimmune thyroid disease, acquired immune deficiency syndrome, and Castleman disease (see Chapter 23 ). Other diffuse interstitial lung diseases encountered in connective tissue diseases, such as diffuse alveolar damage and diffuse pulmonary hemorrhage, are not commonly associated with Sjögren syndrome.
Lung Function
It is difficult to obtain a clear picture of the pattern of abnormal pulmonary function in primary Sjögren syndrome from the literature, and some studies report both restrictive and obstructive abnormalities. However, a 10-year follow-up of pulmonary function in patients with primary Sjögren syndrome found that most patients do not have progressive lung disease.
Manifestations of the Disease
The most frequently observed clinical manifestations of primary Sjögren syndrome are dry mouth, dry eyes, and parotid swelling. The main extraglandular manifestations are arthralgia and myalgia. The range of extraglandular pathology can significantly delay diagnosis in this condition, but in patients with known connective tissue disease, it is important to be alert to symptoms of “sicca syndrome.”
Parenchymal disease: common
- •
Nonspecific interstitial pneumonia
- •
Usual interstitial pneumonia
- •
Lymphocytic interstitial pneumonia
- •
Organizing pneumonia
- •
Lymphoma
- •
Amyloid deposits
- •
Airway disease: common
- •
Follicular bronchiolitis
- •
Bronchiectasis
- •
Vascular disease: uncommon
- •
Pulmonary hypertension
- •
Pleural disease: uncommon
- •
Pleural effusion
- •
Pleural thickening
- •
Radiography
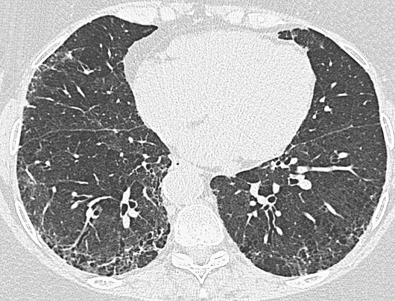
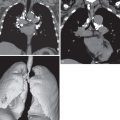
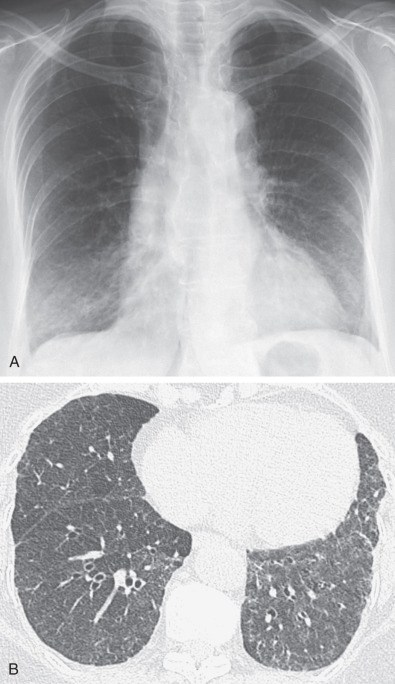
One in 10 patients with primary Sjögren syndrome has pulmonary involvement evident on the chest radiograph. The most common pulmonary manifestations (NSIP, UIP, or LIP) give rise to a reticular or reticulonodular pattern and ground-glass opacities usually involving mainly the lower lung zones ( Fig. 43.1 ). Patients with pure LIP have a reticulonodular pattern on chest radiography, with or without ground-glass opacities and patchy areas of consolidation. The prevalence of chest radiographic abnormalities varies between studies, and in one series, all patients with primary Sjögren syndrome had bilateral consolidation, reticulonodular opacities, or multiple cysts. The majority of these abnormalities occurred in the lower zones. Pleural disease is rare in primary Sjögren syndrome.
Patients with Sjögren syndrome and supervening lymphoma are likely to have abnormal mediastinal and hilar contours on chest radiography, but a normal chest radiograph does not preclude nodal disease. In patients with the rarer primary lymphomas of lung parenchyma or MALT lymphoma, a chest radiograph may show patchy ground-glass opacity and sometimes indolent or, indeed, static areas of consolidation on serial radiographs.
Computed Tomography
Few studies have concentrated exclusively on Sjögren syndrome of the lung, and because its prevalence is approximately equal to that of RA, it can be presumed that Sjögren syndrome either tends to not be associated with lung involvement or runs a subclinical course. The prevalence of HRCT “abnormalities” in those who fulfill the diagnostic criteria is between 34% and 65%, but the majority of patients recruited to these studies had no respiratory symptoms.
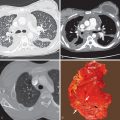
Koyama and colleagues studied 60 patients with abnormal HRCT findings who fulfilled the diagnostic criteria for primary Sjögren syndrome. The most common CT findings were areas of ground-glass opacity (92%), subpleural small nodules (78%), nonseptal linear opacity (75%), interlobular septal thickening (55%), bronchiectasis (38%), cysts (30%), and airspace consolidation (25%). The nodules and septal thickening, although nonspecific, would be consistent with LIP or follicular bronchiolitis ( Fig. 43.2 ). The cysts are also consistent with LIP, and the consolidation could represent lower respiratory tract infection, lymphoproliferative disease, or organizing pneumonia.
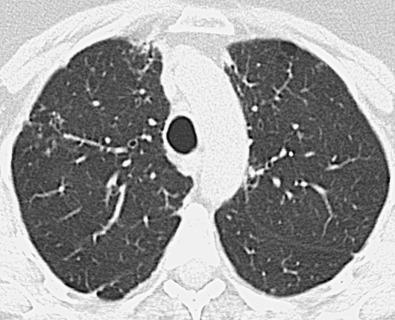
Another study that attempted to characterize the appearances of primary Sjögren syndrome on HRCT recruited nonsmokers, and most of the CT abnormalities were found in those with respiratory symptoms. Similar to the previous study, the most frequent findings on HRCT were ground-glass opacities, linear opacities (reticulation), and airspace consolidation. The histologic correlate for the ground-glass opacities (frequently seen in the various HRCT studies of Sjögren syndrome) is uncertain; NSIP, LIP, infection, and organizing pneumonia are all possible explanations for the ground-glass opacity. The proportion of patients with honeycombing on HRCT varies between 8% and 25%. In Sjögren syndrome there is an increased prevalence of micronodules and interlobular septal thickening, but the pathologic correlate for these lesions is unknown.
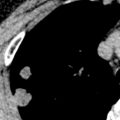
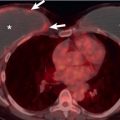
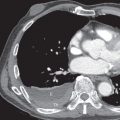
A CT-pathologic correlation in primary Sjögren syndrome found that the NSIP pattern on HRCT had a high positive predictive value (94%) for the presence of NSIP in the pathologic specimen; however, cases without an NSIP pattern correlated poorly with the histologic diagnosis. In this study there were no cases of biopsy-proven LIP despite three patients having cysts on CT; in these three patients, amyloidosis and malignant lymphoma were diagnosed histologically. The HRCT manifestations of NSIP consist of predominantly ground-glass opacities commonly with superimposed reticulation, traction bronchiectasis, and bronchiolectasis and typically involving mainly the lower lung zones (see Fig. 43.1 ). More extensive reticulation, with or without associated honeycombing, may result from NSIP or UIP ( Fig. 43.3 ).