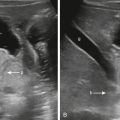
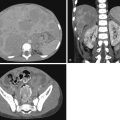
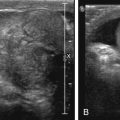
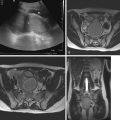
Skeletal dysplasias are bone and cartilage disorders that result in abnormal skeletal development and often, short stature. Skeletal dysplasias and syndromes with bony involvement are not uncommonly seen in pediatric populations. The differential diagnosis of dysplasias includes more than 450 heritable disorders. Early and accurate diagnosis of skeletal dysplasia/syndrome is essential to determining prognosis, receiving appropriate treatment and management of associated complications, and obtaining referral to genetic counseling for future family planning. This chapter will describe a radiological approach to diagnosing skeletal dysplasia using a working algorithm, while illustrating the classic features of many common and visually classifiable skeletal syndromes and dysplasias. Several rare but well-described “Aunt Minnie” diagnoses will be reviewed as well.
When there is a clinical suspicion for a diffuse skeletal syndrome or dysplasia, most evaluations begin with a skeletal survey for osseous abnormalities and a genetics consultation. Dedicated neuroimaging may occur if clinical suspicion for craniosynostosis (computed tomography) or neurological disorder, such as developmental delay or seizures (magnetic resonance imaging). Skeletal surveys performed for evaluation of dysplasias are similar to those for abuse, except the oblique rib radiographs are not performed, and the long bones may be imaged together (femur with tibia/fibula, humerus with radius/ulna, or entire limbs if child is small). Dedicated skull, chest, pelvis, and spine radiographs are also obtained.
Multiple classification systems have been used to characterize skeletal dysplasias; in this chapter, the mnemonic “HELPS ME” will be used to highlight all of the features to evaluate or comment about in the work-up of a skeletal dysplasia, including bones most commonly involved: head (skull), extremities, length, pelvis, spine, mineralization, and extra clinical information. The systematic approach and what to look for follows.
The head should be evaluated on anteroposterior and lateral views of the skull. Analysis should include evaluation of the sutures, including width, evidence of premature fusion or craniosynostosis, and/or presence of a large fontanelle. Specific evaluation for the presence of frontal bossing, Wormian bones (intrasutural bones), midface hypoplasia, and teeth abnormalities should be made. The contour of the skull should be evaluated for lytic lesions, “Lückenschädel” appearance (thinning of the skull), or “copper beaten skull” deformities (gyral impressions seen throughout the skull vault). The sella shape and presence of external auditory canals also should be carefully examined and may be better evaluated on computed tomography.
When evaluating the extremities , the part of the individual long bone that is abnormal should be included in the analysis: the epiphysis, metaphysis, or diaphysis. Look for stippling, delayed ossification, epiphyseal flattening, metaphyseal widening/cupping, bowed diaphyses, and fractures, old and new ( Fig. 11.1 ). Keep in mind that fractures of different ages can be indicative of nonaccidental trauma; however, multiple age-indeterminate fractures are also hallmark features of osteogenesis imperfecta, which is a diagnosis of exclusion.

Simultaneously, the length of the dominant extremity part involved should be evaluated. Portions of the limbs involved can be classified as acromelic (involving the hands or feet), mesomelic (involving the forearm, tibia/fibula), or rhizomelic (involving the humerus or femur). Additional evaluation for extra digits or fusions, radioulnar synostosis, and/or absent bones should be noted. Special attention should also be made for hypoplastic or absent clavicles/scapulae, shortened ribs, and narrow thorax.
The pelvis is also frequently affected by skeletal dysplasias, making observation of the iliac, ischium, pubis, and acetabulum pertinent to the work-up of these syndromes. Certain skeletal dysplasias are associated with pathognomonic “tombstone” iliac bones. The presence of lacy crests or “Mickey Mouse” iliac bones also help narrow the differential diagnosis. Defective development or complete absence of the pubic bones is also characteristic of certain skeletal dysplasias. Trident or horizontal acetabula can offer clues to certain skeletal dysplasias or syndromes as well.
The spine should be carefully evaluated because vertebral anomalies are commonly seen in skeletal dysplasias. The shape of the vertebral bodies (e.g., platyspondyly, bullet-shaped, beaked), presence of fusion or segmentation (i.e., butterfly segmentation) anomalies, as well as agenesis or dysgenesis of the vertebral bodies should be evaluated. Curvatures of the spine should be included in this analysis.
Mineralization or density of the bones should be considered in the work-up of skeletal dysplasias and syndromes. Undermineralization, undertubulation (expanded metaphyses, Erlenmeyer flask deformities), and bone age can give insight into categorization of dysplasias/syndromes as well.
Extra clinical information , such as clinically noted facial dysmorphias, ophthalmological involvement, hearing loss, genetic testing, presence of extraskeletal organ involvement (e.g., cardiac, renal, hepatosplenomegaly, brain malformation), growth/developmental delay, and metabolic/laboratory aberrations, are pertinent for accurate diagnosis and treatment of skeletal dysplasias/syndromes. Consideration of “mimickers” of skeletal dysplasias may fall within the diagnostic differentials as well, including, but not limited to, rickets, scurvy, Blount disease, and physiological bowing.
The following sections will review the most common diagnoses and the rare but classic radiographic appearance of skeletal dysplasias.
Skeletal Dysplasias With Predominant Skull Involvement
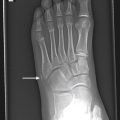
Craniosynostosis is a condition in which the fibrous sutures in the skull prematurely fuse, affecting the growth pattern of the skull. Craniosynostosis is usually isolated to one suture and idiopathic; however, the presence of multiple suture involvement, particularly bilateral coronal craniosynostosis, may suggest syndromic association. The most common syndromes associated with craniosynostosis include Apert, Crouzon, Pfeiffer, Muenke, Saethre-Chotzen, and Antley-Bixler. Most of these conditions are due to mutations on fibroblast growth factor receptor (FGFR) genes, most commonly FGFR2 , with additional genetic mutations in FGFR1 gene in Pfeiffer, FGFR3 in Crouzon and Muenke, and TWIST1 gene in Saethre-Chotzen. Individuals affected by these syndromic disorders typically have exophthalmos and midface hypoplasia. Associated issues include sleep apnea, airway compromise, and hydrocephalus. Several of these are also termed acrocephalosyndactylies because of additional hand/foot malformations. Apert syndrome is also known as type I acrocephalosyndactyly and has an incidence of 1 in 65,000 to 80,000 pregnancies. Patients with Apert syndrome have characteristic soft tissue and osseous syndactyly (“hoof” hand/foot) ( Fig. 11.2 ).

Crouzon syndrome is also characterized by premature craniosynostoses. It has a similar incidence at approximately 1 in 62,500 pregnancies and is associated with a bifid uvula in addition to exophthalmos and midface hypoplasia. It is associated with Chiari type I malformations and cervical spine fusion. It is not frequently associated with limb abnormalities.
Saethre-Chotzen, or type III acrocephalosyndactyly, is the most common craniosynostosis syndrome and affects 1 in 25,000 to 50,000 individuals without predilection for male or female sex. The craniosynostosis typically occurs in the coronal plane, and syndactyly of two or three digits of the hand is typical. There is a characteristic appearance of the ears with a small pinna and prominent superior and/or inferior crus. Individuals with this syndrome also characteristically have ptosis. Cardiac anomalies are associated with Saethre-Chotzen syndrome. Mutations in the TWIST1 gene located on chromosome 7p21 are inherited in an autosomal dominant pattern and lead to this characteristic syndrome.
Pfeiffer syndrome, or type V acrocephalosyndactyly, is characterized by proptosis, brachydactyly (particularly broad thumbs/great toes), and maxillary hypoplasia in addition to craniosynostosis and syndactyly. It arises from mutations in the FGFR1 gene on chromosome 8 or the FGFR2 gene on chromosome 10. There are three types of Pfeiffer syndrome, with type 1 being the classic syndromal appearance, with individuals having normal intelligence; types 2 and 3 have poorer prognosis with severe neurological compromise and proptosis, respectively.
Severe multisutural fusions can cause a “cloverleaf” skull deformity, which has been associated with many syndromes, including Crouzon, Pfeiffer, and Carpenter, as well as type II thanatophoric dysplasia.
Cleidocranial dysostosis is a skeletal dysplasia with distinct skull involvement and is characterized by incomplete intramembranous ossification of certain skeletal structures. Cranial manifestations include multiple Wormian bones, frontal bossing, brachycephaly, high arched palate, and supernumerary teeth ( Fig. 11.3 ). The clavicles and pubic bones are often shortened or absent. The vertebral column (mild and long bones) may also be affected. The thorax is often affected with a narrowed, bell-shaped appearance, which can lead to respiratory distress in severe cases. There is abnormal development of the ear structures, which can cause hearing loss in some cases. A majority of patients with cleidocranial dysostosis have a mutation in the RUNX2 gene on chromosome 6, with an autosomal dominant inheritance pattern. Approximately 30% of patients do not have an identified causal mutation.

Pyknodysostosis, or Toulouse-Lautrec syndrome, is characterized by osteosclerosis and short stature. Patients present with delayed closing of cranial sutures and frontal/occipital bossing, short broad hands, and multiple long bone fractures after minimal trauma. It is a lysosomal disorder due to a deficiency of cathepsin K, which is necessary for normal osteoclast function. Radiographically, there is evidence of delayed bone age, aplasia of the terminal phalanges, Wormian bones, proptosis, prognathism, vertebral segmentation anomalies (primarily upper cervical and lower lumbar), and hypoplastic clavicles. The abnormality is on the CTSK gene on chromosome 1 and is inherited in an autosomal recessive pattern.
Multiple syndromes involve facial development, such as Goldenhar syndrome; although sometimes referred to as hemifacial microsomia, it may manifest as a bilateral condition and is a severe manifestation along the oculo-auriculo-vertebral spectrum characterized by abnormalities of the eyes, ears, and vertebrae. Patients manifest with underdevelopment of the affected facial bones with otic hypoplasia, preauricular appendages, and ipsilateral microphthalmia. It occurs sporadically with a slight male predominance and an incidence of 1 in 3000 to 5000 births.
Facial underdevelopment is Pierre Robin sequence, which is characterized by mandibular micrognathia, glossoptosis (retraction of the tongue), and a high or U-shaped palate. Severe micrognathia may lead to airway obstruction and hypoxia. Robin sequence occurs in 1 in 8500 births. DNA mutations near the SOX9 gene are thought to be the inciting genetic cause of the disorder. Nongenetic causes, such as oligohydramnios and in utero restriction, are speculated to result in isolated Pierre Robin sequence because of jaw growth restriction.
Extremities: Dysplasias With Prominent Diaphyseal Involvement
Thanatophoric dysplasia is the most common lethal skeletal dysplasia with an incidence of 1 in 25,000 to 50,000 births. It results from a mutation in the FGFR3 gene on chromosome 4p16 and occurs sporadically. There are two subtypes: type I is associated with “telephone receiver”–shaped femurs ( Fig. 11.4 ), while type II is characterized by a “cloverleaf skull” and milder limb shortening. The proximal long bones are typically shortened, giving a rhizomelic appearance, with bowing and metaphyseal flaring. The iliac bones are hypoplastic, and there may be “trident acetabula,” as well as platyspondyly. The thorax may be narrowed with shortened ribs and small scapulae. The condition is lethal within a few hours of birth secondary to respiratory failure or brainstem compression from a narrowed foramen magnum.

Bent bone dysplasias predominantly affect the diaphyses. They are syndromes characterized by lower extremity bowing, hypoplastic scapulae, dysplastic acetabula and iliac bones, and dislocated hip and knee joints. Campomelic dysplasia, meaning “bent limb” in Greek, is a bent bone dysplasia characterized by a hypoplastic mandible, cephalad angulation of the clavicles, hypoplastic scapulae, and rib anomalies (i.e., 13 pairs) ( Fig. 11.5 ). It is attributed to a mutation in the FGFR2 gene and has an incidence of 1 in 200,000 births with an autosomal recessive inheritance. It is a relatively lethal disorder, causing death in up to 97% of patients within the first year of life caused by respiratory failure.