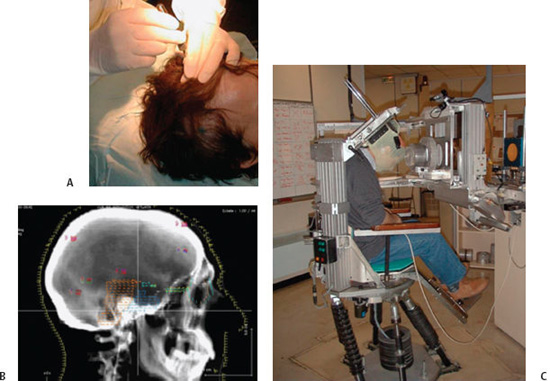
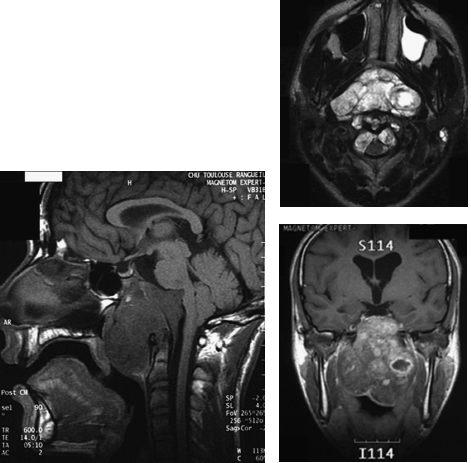
10 Skull base chondrosarcomas and chordomas are very rare neoplasms that share similarities that justify a joint study on these entities: both can be located in the skull base or along the spinal axis; both share an indolent course in the majority of cases; both are highly invasive locally; and in the long run, a fatal outcome is inevitable if they are not adequately managed. From a therapeutic standpoint, both are formidable challenges for neurosurgeons and radiation oncologists, due to their invasiveness, proximity to vital structures, and sensitivity to therapeutic irradiation. Nonetheless, considerable progress has been achieved over the past decade or so in the outcome of both tumor types, due to the development of safer extensive skull base resections, combined with innovative radiosurgery and radiotherapy approaches, especially proton therapy. This chapter focuses on this latter technique. Chondrosarcomas are very rare neoplasms that account for 0.5 case per million1 and ~1 per 1000 malignancies in the head and neck region. They generally occur in the 2nd and 3rd decade of life with no predilection for gender. Anatomically, the skull base ranks generally behind more common presentations in the nasal/paranasal cavities, the mandible, or the orbits. Within the skull base, they tend to arise more frequently off the midline in the petrous bone, parasellar area, or cerebellopontine angle. Macroscopically, they frequently present as large lesions destroying existing bone. Microscopically, they have been divided into “conventional,” mesenchymal, clear cell, and “dedifferentiated” variants (World Health Organization classification).2 The common low-grade variant exhibits a lobulated appearance with frequent central necrosis and calcifications, associated with well-differentiated hyaline cartilage, and a myxoid ground substance. In higher grade variants, cartilage architecture is generally lost and associated with increased cellularity, mitoses, and larger nuclear sizes. Grading exerts a considerable prognostic impact. Indeed, 50 to 60% of chondrosarcomas are classified as low-grade (grade 1) lesions based on the nuclear appearance and cellularity, with the remaining lesions classified as grade 2 or 3. The very rare “mesenchymal” and “undifferentiated” variants include, respectively, highly undifferentiated small round cells and high-grade noncartilaginous sarcomas. Immunohistochemically, most of them display positive staining for mesenchymal antigens (vimentin) and sometimes for neuroepithelial antigens (S100 protein) and glial fibrillary acidic protein (GFAP). These features can help differentiate other low-grade malignancies, such as chordomas. More recently, genetic alterations have been described in association with chondrosarcomas: they consist of alterations of genes involved in tumor suppression, proliferation regulation, cell cycle regulation and differentiation, and repair. They are located on chromosomes 5, 9, 11, 13, 17, and 19. Chondrosarcomas can be either primary or secondary lesions, depending on whether they arise in a previously normal bone or in a benign precursor, such as enchon-droma. The risk of malignant transformation is quite low in cases of isolated foci but greater in cases of widespread congenital enchondromatoses: the multiple enchondromatosis (Ollier disease) carries a 25 to 30% risk of developing into secondary mesenchymal malignancies; the Mafucci syndrome, which combines Ollier bony anomalies and soft tissue angiomas, carries a 25 to 50% risk of developing into secondary polymorphic malignancies. Chordomas are also very rare neoplasms, found in < 1% of autopsy-verified central nervous system (CNS) tumor processes, representing ~1 case per million.1 The mean age is ~45 to 50 years, with cases described as early as 3 years and as late as 80 years, and a predilection is seen for males. They likely originate from remnants of the notochord, an embryonic structure located along the craniovertebral axis, which serves as primitive backbone support of the body and regresses by the 6th week of development. Their jelly-like appearance led early scientists to postulate links with the intervertebral disk, known as the nucleus pulposus. More recently, the role of the notochord in the embryonic formation of the dorsal and ventral roots of the spinal cord was emphasized.3 Chordomas can arise anywhere along the primitive notochord, with ~45 to 50% of cases described in the sacrum, 35 to 40% in the skull base, and 15% along the spine. Anatomically, intracranial sites affect the midline, especially the spheno-occipital junction, but “aberrant” locations (i.e., sphenoid, maxillary, and frontal sinuses) have been reported. Macroscopically, chordomas are lobulated in appearance with gelatinous processes. Histologically, cells are arranged in sheets or cords or float singly within an abundant myxoid stroma (that can be confused with myxoid chondrosarcoma). Typically, they contain abundant pale, vacuolated cytoplasm (“physaliphorous”) and moderate nuclear atypias. In the chondroid variant, which has the reputation of a more indolent course, some areas may mimic hyaline or myxoid cartilage, a feature likely to cause confusion with true chondrosarcomas. The dedifferentiated (including high-grade sarcoma areas) and “highly cellular” variants are extremely rare and distinctly aggressive. By immunohistochemistry, most of them display polymorphic positive stainings for epithelial (cytokeratin, epithelial membrane antigen), neuroepithelial (S100 protein), and mesenchymal (vimentin) antigens. Genetic alterations consist of gains in chromosomes 5, 7, 12, and 20 and losses of 1 and 3. These have been linked to alterations of tumor suppressor genes, mismatched repairs, and oncogenes. Symptoms are generally insidious and nonspecific. Half of the patients present with ocular symptoms linked to oculomotor palsy (i.e., diplopia) or compression of the visual pathway (bitemporal hemianopia). Headaches, which occur typically in the retro-orbital and occipital regions, are also common at presentation. Snoring can be reported when a nasopharyngeal or pharyngeal mass is present. These symptoms are frequently neglected by the clinicians and the patients themselves. Generally, they require specific imaging after several years while they have been deteriorating. Radiologically, computed tomography (CT) scans and magnetic resonance imaging (MRI) are the most useful modalities for assessing tumor extension. Chondrosarcomas appear on CT scans as large off-midline lesions destroying bone, with calcifications and rather well-defined margins. On MRI, the appearance is generally hypointense on T1-weighted images and strongly hyperintense on T2-weighted images. They usually enhance contrast medium poorly due to their hypovascu-larity. For chordomas, CT imaging is superior for detecting bony destruction, whereas MRI is useful for assessing the diagnosis and for defining tumor extension; typically, the tumor displays a lobulated, “honeycomb” appearance on contrast-enhanced images and is located in the clival area, which displaces or even distorts the brainstem posteriorly and the optic pathway superiorly1,4 (Fig. 10.1). On T1-weighted images, the tumor demonstrates an intermediate to low signal with foci of hyperintensity that can represent residual osseous fragments, calcifications, or proteinacous or hemorrhagic fluid. On T2-weighted images, it demonstrates very high signal intensity arranged with low signal septa. Chordomas usually enhance the contrast medium slightly. The evolution of both diseases is generally slow although invariably lethal if not appropriately managed. Following therapy, imaging is crucial to assess tumor control. The radiologic aspects remain frequently stable for months or years and should not be interpreted as local failure. The malignant nature of chordomas is supported by the reports of distant hematogenous foci (nodal, pulmonary, hepatic, cutaneous, etc.), not to mention second synchronous or metachronous primaries located within the clival bone or in the CNS. Failures have been described along the surgical route, as well as within the abdominal wall where fat was removed during surgery.5,6 Skull base chordomas and chondrosarcomas require advanced radiosurgery or radiotherapeutical technologies capable of delivering high-dose, high-precision radiation. Although radiosurgery can be used, this chapter will focus on proton therapy for treating skull base sarcomas. A considerable tumor burden generally persists after surgery. Both diseases are notorious for being highly radioresistant, and they both frequently abut sensitive anatomical structures (e.g., optic nerve, chiasm, brainstem, and spinal cord) whose injury could be lethal or highly damaging. Dose fractionation may provide biological advantages in terms of tumor sensitivity and normal organ repair mechanisms. These are the numerous reasons why proton therapy has become a major therapeutical tool since the mid-1970s. Physical and Biological Properties In contrast with photon beams, which are exponentially absorbed in matter, charged particles, such as electrons and protons, exhibit a finite range. Because the mass of protons is 2000-fold that of electrons, they carry considerably higher kinetic energy (at equal velocity); therefore, a vastly greater release of energy is obtained as they are slowing down. Furthermore, these “projectiles” are barely deflected during the process, which translates into a sharp lateral “penumbra” on dose profiles.7 Monoenergetic proton beams generated in large accelerators such as synchrotrons produce particles exhibiting a highly uniform range (roughly 1%) and deep penetration (~15–30 cm in water-equivalent material). These properties translate into the so-called Bragg curve: (1) relatively limited interactions in their initial path and limited dose-absorption and (2) a steep rise toward the end of their range, followed by a sharp fall-off of radiation dose. A slight increase in the relative biological effectiveness (RBE) along the beam’s path that requires appropriate correction of the physical dose (by 10%) has been reported.8 The “pristine” Bragg peak does not exceed a few millimeters at midheight and needs to be “spread out” to encompass thick lesions. The drawback, however, is an increase in the entrance dose. In practice, this disadvantage can be overcome by using multiple angularly separated beams, which is facilitated by using an isocentric gantry, and by technologies under development, such as spot scanning. The latter approach, which is available in the Paul Scherrer Institute, Villigen, Switzerland, improves the modulation of each field by delivering actively modulated small “beamlets.”9 It should be pointed out that the stopping power of different tissues for protons differs substantially as a function of their density; for example, major corrections are required when the beam passes from bones to soft tissues (i.e., water equivalent) and even more to air cavities. These differences can be “compensated” for by interposing Lucite “compensators” within the beam path in the passive scattering approach or by using the more recent spot-scanning technology without an additional compensator. Technical Aspects Early proton facilities were established on university campuses (e.g., centre de Protonthérapie d’Orsay, Orsay, France) or in nuclear research centers (e.g., the Paul Scherrer Institute). These facilities featured simple technology—fixed beam, passive scattering, handmade compensators, for example.10 Recently, hospital-based facilities have been developed, especially in the United States and Japan. This became possible because modern accelerators are somewhat more compact than their earlier counterparts. Although synchrotrons seem to be the most appropriate accelerators for producing protons of high (i.e., 200–250 MeV) and variable energies, recently interest has been directed to modified cyclotrons, which can achieve excellent performances at relatively low cost (“isochronous” cyclotrons). Newer technology features sophisticated beam delivery systems, including isocentric gantries that help set up complex beam arrangements but are still considerably space consuming. Approximately 25 centers worldwide have implemented a clinical program. Patient Setup and Alignment We will briefly discuss the procedure implemented at the Orsay center. The patient can be lying supine or seated (Fig. 10.2). A thick custom-made thermoplastic mask is used for immobilization purposes. A bite block can be used concomitantly in compliant patients to avoid head tilt movements. Final accuracy largely depends on stereotactic alignment, based on the implantation of four or five radiopaque nonfer-romagnetic fiducial markers in the outer skull under local anesthesia. Orthogonal x-ray films are compared with digitally reconstructed radiographs before each daily session to check alignment and repeated for each field. The fiducial markers allow appropriate translation and angular corrections using an original computer program (ROTAPLUS) developed at the Institut Curie d’Orsay Proton Therapy Center. Appropriate corrections are made using a high-precision robotic chair or a couch. Setup accuracy has been verified to < 1 mm in the X, Y, and Z directions and 1 degree for the rotation parameter. Proton therapy treatment time averages 25 minutes, with 20 minutes for the patient setup and 5 minutes for irradiation itself. Possible displacement during the irradiation session is minimized by the visualization of magnified landmarks drawn on the mask or on a TV monitor or of special external marks on an infrared camera. Most treatments are conducted in a conventionally fractionated way (i.e., 30 to 40 daily fractions of 2 CGE cobalt gray equivalent [CGE] = physical dose in Gy + 10% dose for RBE). Treatment Planning and Simulation Treatment planning and simulation are conducted using high-definition fused CT-MRI images recorded in the treatment position. Three-dimensional (3D) treatment plans, including dose–volume histograms, are generated using “homemade” software (ISIS) recently upgraded by the DO-SIsoft (Cachan, France). Proton beam arrangement generally combines six or seven noncoplanar beams that can be matched using both their lateral and distal penumbras (beam “patching”) (Fig. 10.3). In adults, approximately half of the treatment is delivered using photons and the other half with protons (Fig. 10.4). This does not seem to compromise tumor coverage compared with protons alone.11 Children are exclusively treated with protons to maximize the shielding of normal structures even in the “low” and “medium” dose regions. Fig. 10.2 Patient’s preparation and setup before fractionated proton beam therapy. (A) Implantation of intracranial fiducial markers, (B) digitally reconstructed radiographs used for alignment, and (C) patient with thermoplastic mask in robotic chair.
Stereotactic Radiosurgery for Skull Base Chordomas and Chondrosarcomas
Jean-Louis Habrand, Stéphanie Bolle, Régis Ferrand, and Georges Noel
 Epidemiology and Pathogenesis
Epidemiology and Pathogenesis
Chondrosarcomas
Chordomas
 Patient Presentation
Patient Presentation
 Management
Management
Proton Therapy
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


