Etiology
Systemic sclerosis (scleroderma) has three cardinal features: excessive collagen production, vascular damage, and inflammation/autoimmunity. The pathogenesis of systemic sclerosis is obscure, but there are several contributing factors, including genetic, environmental, and autoimmune influences. Familial aggregation for systemic sclerosis, although infrequent (1.2%–1.5% of families), has been established and lays a firmer foundation for genetic studies of its etiopathogenesis.
Systemic sclerosis is considered to be an autoimmune process, possibly resulting from deleterious effects of cytokines on extracellular matrix proteins and tissue healing. Some studies have also implicated cellular microchimerism in its pathogenesis. Microchimerism results from persistence of fetal cells in the maternal circulation from previous pregnancies. The demonstration of fetal CD3 + T cells in the maternal circulation and fetal cells in affected tissues suggests that microchimerism may, in some patients, initiate a graft-versus-host–like response that results in systemic sclerosis.
Prevalence and Epidemiology
Systemic sclerosis is uncommon, with an annual incidence of 20 per million population per year and an estimated prevalence of 500 cases per million population. The incidence of systemic sclerosis increases with age, and it most frequently affects women in their childbearing years. There is no overall racial predilection. Systemic sclerosis is associated with significant mortality, and approximately a third of patients die within 10 years of diagnosis. Variables that best predict reduced survival include older age (>64 years), reduced renal function, anemia, reduced gas diffusing capacity for carbon monoxide (DLCO), reduced total serum protein level, and decreased pulmonary reserve (forced vital capacity). Individuals with anticentromere antibody (ACA) have a better prognosis than those with anti–topoisomerase I (Scl-70) antibody. The poorer prognosis of those with anti–Scl-70 probably relates to the fact that fibrotic lung disease is more likely to develop in these patients and to be manifested as diffuse systemic sclerosis.
Clinical Presentation
Only 1% of patients initially have respiratory symptoms, usually shortness of breath on exertion, but in established disease, dyspnea is the most common pulmonary symptom in systemic sclerosis and occurs in about 60% of patients. Approximately 16% of patients with systemic sclerosis have pleuritic chest pain at some time in their disease. Unlike patients with rheumatoid arthritis or systemic lupus erythematosus, clinically significant pleural effusion rarely develops in patients with systemic sclerosis. Nevertheless, pleural fibrosis and adhesions are frequently identified at autopsy.
Approximately 41% of patients with systemic sclerosis have pulmonary hypertension, and dyspnea on exertion is usually the first symptom of pulmonary hypertension. The risk for pulmonary hypertension in patients with systemic sclerosis increases with the age at disease onset ; gas diffusing capacity may be significantly decreased for many years before the diagnosis of pulmonary hypertension is made (perhaps indicating subclinical disease).
Cardiac symptoms reflect a wide range of possible involvement: right-left ventricular dysfunction (fatigue), pulmonary hypertension (dyspnea), congestive heart failure (edema), pericarditis/angina (chest pain), autonomic dysfunction (palpitations), and rhythm disturbances and conduction defects (dizziness, syncope, sudden death).
Gastrointestinal symptoms, particularly swallowing difficulties, are common. The severity of esophageal motor involvement in systemic sclerosis has been shown to correlate with the severity of pulmonary manifestations of the disease (quantified by decreased DLCO) and evidence of interstitial lung disease on high-resolution computed tomography (HRCT). This may be due to the fact that there is a relationship between gastroesophageal reflux and interstitial lung disease, but an alternative explanation is that they both reflect the severity of the systemic vasculopathy. Systemic sclerosis is accompanied by joint pain in 15% of patients and by inflammatory myopathy in 10% of patients. Muscle weakness, cutaneous pruritus, and Raynaud phenomenon are common. Renal failure can be manifested as hypertensive/normotensive renal failure or as an abrupt onset of a renal emergency.
Scleroderma can be localized to the skin or be systemic. Systemic disease is further divided into limited cutaneous and diffuse cutaneous types. Limited cutaneous systemic sclerosis encompasses the now outdated term “CREST variant” ( c alcinosis, R aynaud disease, e sophageal dysmotility, s clerodactyly, and t elangiectasia). Limited cutaneous systemic sclerosis is defined by skin thickening in areas distal to the elbows and knees, with or without facial scleroderma. By contrast, diffuse cutaneous systemic sclerosis is defined by the presence of skin thickening proximal as well as distal to the elbows and knees, with or without facial or truncal disease. Although lung involvement occurs more frequently in the diffuse form of systemic sclerosis, it is evident in approximately a third of patients with limited cutaneous disease.
Pathophysiology
Pathology
Scleroderma is characterized by fibrosis and vascular obliteration of the skin, gastrointestinal tract, lungs, heart, and kidneys. The lungs are the second most common site of visceral involvement, and pulmonary disease is a major cause of death in patients with systemic sclerosis.
The two main pulmonary manifestations are interstitial fibrosis and pulmonary hypertension; the latter reflects a primary vasculopathy, but it may be secondary to cardiac or pulmonary disease. A postmortem study has reported that three quarters of patients with systemic sclerosis had interstitial fibrosis. Pulmonary fibrosis has been estimated to ultimately develop in 80% of patients with systemic sclerosis and pulmonary arterial hypertension in 50% of cases. Patients with systemic sclerosis generally fall into one of two categories: those in whom fibrotic lung disease develops and who have anti–Scl-70 antibodies (and in whom pulmonary hypertension may develop secondary to their heart or lung disease) and those who have solely pulmonary hypertension and positive serology for ACA. Of interest, ACA and anti–Scl-70 antibodies are almost always mutually exclusive, being simultaneously present in less than 0.5% of patients with systemic sclerosis. Patients with diffuse cutaneous systemic sclerosis and anti–Scl-70 antibody are at especially high risk for fibrotic lung disease.
The most frequent histologic subtype of pulmonary fibrosis is nonspecific interstitial pneumonia (NSIP). A review of lung biopsy specimens taken from patients with systemic sclerosis found that more than 75% of this cohort had NSIP on histologic review. The high frequency of NSIP in this study can probably be extrapolated to a wider population of patients with systemic sclerosis who have not undergone lung biopsy. The prevalence of usual interstitial pneumonia (UIP) in the systemic sclerosis population has been estimated to be less than 10%. There are case reports of diffuse alveolar damage in systemic sclerosis, but it is rare. A few cases of diffuse alveolar hemorrhage have also been reported; in most instances the diffuse alveolar hemorrhage occurs in the setting of preexisting fibrotic lung disease.
Cardiac involvement is virtually always present to some degree in the form of patchy myocardial fibrosis, sometimes causing ventricular diastolic dysfunction. Ventricular systolic dysfunction is also seen in patients with concomitant coronary or hypertensive heart disease. Patients with systemic sclerosis are prone to the development of conduction defects and autonomic cardiac neuropathy.
In a postmortem series, the esophagus was involved in 75% of patients with systemic sclerosis, and at autopsy most organs show evidence of patchy fibrosis or atrophy, or both.
Lung Function
In systemic sclerosis, a diffusing capacity (DLCO) of 50% or less of predicted and reduced spirometric reserve predict reduced survival. Diminished diffusing capacity is the earliest detectable pulmonary function abnormality in patients with systemic sclerosis, and it correlates with clinical symptoms of dyspnea and the presence of pulmonary fibrosis. The diffusing capacity can be reduced because of pulmonary fibrosis or vasculopathy. Reduced diffusing capacity is the best predictor of the extent of fibrotic lung disease in systemic sclerosis.
Manifestations of the Disease
The hallmark of systemic sclerosis is thickened skin. In diffuse scleroderma with facial involvement, a cardinal sign is exaggerated radial furrowing around the lips (tobacco pouch sign). Skin thickening starts with the fingers and hands in virtually all cases; progression of the skin tightening and thickening is variable. Calcinosis is seen over the periarticular surfaces of the hands, elbows, and knees. Telangiectases occur on the hands, face, lips and gastrointestinal mucosal surfaces. The cutaneous macules are a harmless cosmetic problem, but those in the gastrointestinal tract can bleed and lead to iron deficiency anemia. Patients with systemic sclerosis commonly have Raynaud phenomenon (reversible vasomotor instability precipitated by cold or emotion). It is usually manifested as changes in color of the extremities (pallor to cyanosis and eventually erythema).
Radiography
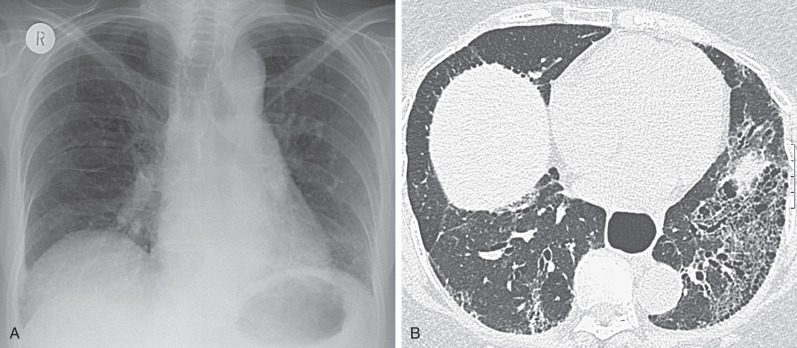
The most frequent abnormality on chest radiography is a widespread, symmetric basal reticulonodular pattern ( Fig. 40.1 ). Depending on the severity of the pulmonary fibrosis, honeycombing, reduced lung volumes, and traction bronchiectasis can be appreciated on the chest radiograph. However, as with all interstitial lung disease, chest radiography lacks the sensitivity of HRCT for the detection of limited parenchymal disease. A dilated air-filled esophagus can be identified in many patients (see Fig. 40.1 ). Because there is no anatomic obstruction, a fluid level is not usually a feature on erect chest radiographs.
Computed Tomography
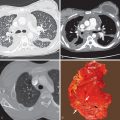
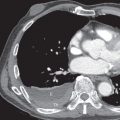
The majority of patients with fibrosing lung disease and systemic sclerosis have a histologic pattern of NSIP rather than UIP ( Fig. 40.2 ; see Fig. 40.1 ). Evidence for this comes from both histopathologic and radiologic studies.
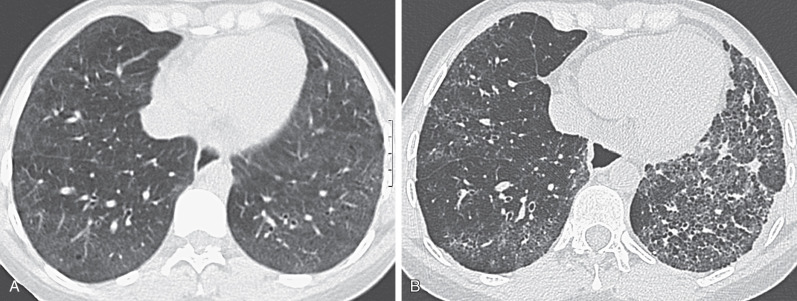
The typical HRCT appearances of NSIP consist of predominantly ground-glass opacities involving mainly the lower lung zones and often mainly the subpleural regions, reticulation, architectural distortion (traction bronchiectasis and volume loss), and occasional limited honeycombing. Although ground-glass opacities may initially be the predominant or only abnormality, over time, progressive reticulation, traction bronchiectasis, traction bronchiolectasis, and, less commonly, areas of honeycombing tend to develop (see Fig. 40.2 ). In many patients traction bronchiectasis is a predominant abnormality. In patients with symptomatic pulmonary involvement in systemic sclerosis, pulmonary fibrosis and ground-glass opacity are common findings, with honeycombing seen more frequently in the limited than the diffuse cutaneous form of systemic sclerosis subtypes. The extent of involvement at HRCT correlates with patient prognosis in systemic sclerosis; in an important study, patients with less than 20% involvement of the lungs had an average 10-year survival of 67%, whereas a group with greater than 20% involvement had an average survival of 43%.
Lymphadenopathy is frequent in patients with systemic sclerosis and interstitial lung disease, and the degree of lymph node enlargement is related to the extent, but not the type, of interstitial disease.
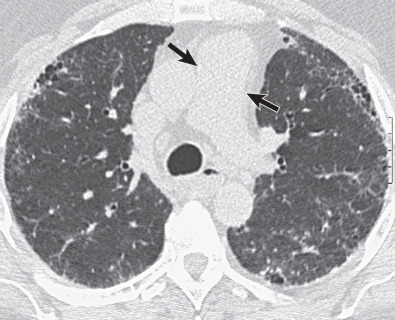
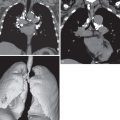
In patients with isolated pulmonary arterial hypertension (and limited scleroderma), evidence of raised pulmonary artery pressure may be a diameter of the main pulmonary artery greater than 3.2 cm, or a diameter larger than that of the adjacent ascending aorta ( Fig. 40.3 ) ; however, a nondilated pulmonary artery does not necessarily exclude pulmonary hypertension.