-amyloid deposition begins in Alzheimer’s disease (AD) years before the onset of any clinical symptoms. It is therefore important to determine the temporal trajectories of amyloid deposition in these earliest stages in order to better understand their associations with progression to AD. A method for estimating the temporal trajectories of voxelwise amyloid as measured using longitudinal positron emission tomography (PET) imaging is presented. The method involves the estimation of a score for each subject visit based on the PET data that reflects their amyloid progression. This amyloid progression score allows subjects with similar progressions to be aligned and analyzed together. The estimation of the progression scores and the amyloid trajectory parameters are performed using an expectation-maximization algorithm. The correlations among the voxel measures of amyloid are modeled to reflect the spatial nature of PET images. Simulation results show that model parameters are captured well at a variety of noise and spatial correlation levels. The method is applied to longitudinal amyloid imaging data considering each cerebral hemisphere separately. The results are consistent across the hemispheres and agree with a global index of brain amyloid known as mean cortical DVR. Unlike mean cortical DVR, which depends on a priori defined regions, the progression score extracted by the method is data-driven and does not make assumptions about regional longitudinal changes. Compared to regressing on age at each voxel, the longitudinal trajectory slopes estimated using the proposed method show better localized longitudinal changes.
1 Introduction
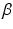 -amyloid deposition is a neuropathological hallmark associated with Alzheimer’s disease (AD), and begins years before any cognitive symptoms of AD are evident [8]. Studying within-subject longitudinal changes is limited by the number of follow-up visits. The relatively short span of longitudinal positron emission tomograpy (PET) studies of amyloid deposition compared to its hypothesized timeline makes it difficult to extensively study the longitudinal brain amyloid changes that occur in the preclinical stages of AD.
-amyloid deposition is a neuropathological hallmark associated with Alzheimer’s disease (AD), and begins years before any cognitive symptoms of AD are evident [8]. Studying within-subject longitudinal changes is limited by the number of follow-up visits. The relatively short span of longitudinal positron emission tomograpy (PET) studies of amyloid deposition compared to its hypothesized timeline makes it difficult to extensively study the longitudinal brain amyloid changes that occur in the preclinical stages of AD.2 Method
2.1 Model
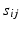 for subject i at visit j is assumed to be a linear transformation of age
for subject i at visit j is assumed to be a linear transformation of age 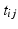 :
: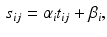
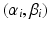 are assumed to be uniformly distributed random variables on a fixed and large domain
are assumed to be uniformly distributed random variables on a fixed and large domain 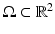 . The collection of K biomarker measurements (the intensities at each PiB-PET DVR voxel reflecting amyloid levels) make up the
. The collection of K biomarker measurements (the intensities at each PiB-PET DVR voxel reflecting amyloid levels) make up the 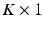 vector
vector 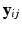 . The longitudinal trajectories associated with these biomarkers are assumed have a linear form parameterized by
. The longitudinal trajectories associated with these biomarkers are assumed have a linear form parameterized by 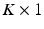 vectors
vectors ![$$\mathbf {a} = [a_1, a_2, \ldots , a_K]^T$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_IEq10.gif) and
and ![$$\mathbf {b} = [b_1, \ldots , b_K]^T$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_IEq11.gif) :
: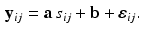
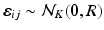 is the observation noise. The covariance matrix R is assumed to be of the form
is the observation noise. The covariance matrix R is assumed to be of the form 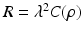 , where
, where 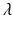 is a positive scalar and C is a correlation matrix parameterized by
is a positive scalar and C is a correlation matrix parameterized by 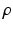 . The parameters
. The parameters 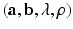 make up
make up 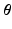 .
.2.2 Log-Likelihood Function
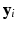 be the vector consisting of all biomarker measurements stacked across all visits of subject i and let
be the vector consisting of all biomarker measurements stacked across all visits of subject i and let ![$$\mathbf {u}_i = [\alpha _i, \beta _i]^T$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_IEq19.gif) . We then define
. We then define ![$$\mathbf {y} = \left[ \mathbf {y}^T_1, \mathbf {y}^T_2, \ldots , \mathbf {y}^T_n\right] ^T$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_IEq20.gif) and
and ![$$\mathbf {u} = \left[ \mathbf {u}^T_1, \mathbf {u}^T_2, \ldots , \mathbf {u}^T_n\right] ^T$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_IEq21.gif) , where n is the number of subjects. The pair
, where n is the number of subjects. The pair 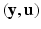 is the complete data. The complete log-likelihood for the model, ignoring the constants, is given by
is the complete data. The complete log-likelihood for the model, ignoring the constants, is given by![$$\begin{aligned} \ell (\mathbf {y}, \mathbf {u} \mid \varvec{\theta })= & {} -\sum _{i,j} \left[ \log (\det \, R ) + \left( \mathbf {y}_{ij} - Z_{ij} \mathbf {u}_i - \mathbf {b} \right) ^T R^{-1} \left( \mathbf {y}_{ij} - Z_{ij} \mathbf {u}_i - \mathbf {b} \right) \right] , \; \; \; \; \end{aligned}$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_Equ3.gif)
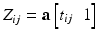 and
and 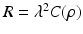 .
.2.3 EM Algorithm Derivation
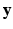 include biomarker measurements
include biomarker measurements 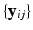 at each visit. The hidden variables
at each visit. The hidden variables 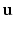 are the subject-specific parameters
are the subject-specific parameters 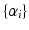 and
and 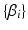 . The unknown parameters are
. The unknown parameters are 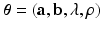 .
.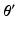 be the previous estimate of the parameters. The expectation step involves evaluating an expression for
be the previous estimate of the parameters. The expectation step involves evaluating an expression for![$$\begin{aligned} E \left[ \ell \left( \mathbf {y}, \mathbf {u} \mid \varvec{\theta } \right) \mid \mathbf {y}, \varvec{\theta }' \right]= & {} \sum _i \int f(\tilde{\mathbf {u}}_i \mid \mathbf {y}_i, \varvec{\theta }') \ell \left( \mathbf {y}_i, \tilde{\mathbf {u}}_i \mid \varvec{\theta }\right) d\tilde{\mathbf {u}}_i. \end{aligned}$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_Equ4.gif)
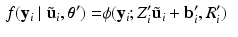
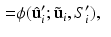
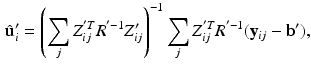
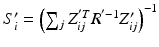 , and
, and 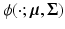 is a multivariate Gaussian density with mean
is a multivariate Gaussian density with mean 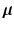 and covariance
and covariance 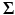 . For ease of notation, we have used
. For ease of notation, we have used 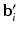 to denote
to denote 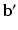 stacked across the visits of subject i and
stacked across the visits of subject i and 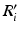 to denote diagonally stacked
to denote diagonally stacked 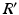 matrices across the visits of subject i.
matrices across the visits of subject i. 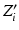 is obtained by diagonally stacking
is obtained by diagonally stacking 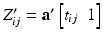 across the visits of subject i.
across the visits of subject i.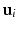 allows us to write
allows us to write 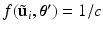 , where c is a constant. Rewriting the expectation using Bayes’ rule and plugging in the result from (6) yields
, where c is a constant. Rewriting the expectation using Bayes’ rule and plugging in the result from (6) yields![$$\begin{aligned} E \left[ \ell \left( \mathbf {y}, \mathbf {u} \mid \varvec{\theta }\right) \mid \mathbf {y}, \varvec{\theta }' \right]\propto & {} \sum _i \int _\Omega \frac{\phi (\hat{\mathbf {u}}'_i; \tilde{\mathbf {u}}_i, S'_i)}{f(\mathbf {y}_i,\varvec{\theta }')} \ell \left( \mathbf {y}_i, \tilde{\mathbf {u}}_i \mid \varvec{\theta }\right) d\tilde{\mathbf {u}}_i. \end{aligned}$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_Equ8.gif)
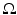 is large enough, we can approximate the expectation as an infinite domain integration over
is large enough, we can approximate the expectation as an infinite domain integration over 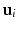 to obtain
to obtain![$$\begin{aligned} E \left[ \ell \left( \mathbf {y}, \mathbf {u} \mid \varvec{\theta }\right) \mid \mathbf {y}, \varvec{\theta }' \right]\propto & {} \sum _i \int \phi (\hat{\mathbf {u}}'_i; \tilde{\mathbf {u}}_i, S'_i) \ell \left( \mathbf {y}_i, \tilde{\mathbf {u}}_i \mid \varvec{\theta }\right) d\tilde{\mathbf {u}}_i \\\propto & {} -\sum _{i,j} \log [\det R] -\sum _{i,j} (\mathbf {y}_{ij} - \mathbf {b})^T R^{-1} (\mathbf {y}_{ij} - \mathbf {b}) \nonumber \\&+ 2 \left( \sum _{i,j} s'_{ij} \mathbf {y}_{ij} - \mathbf {b} \sum _{i,j} s'_{ij} \right) ^T R^{-1} \mathbf {a} \nonumber \end{aligned}$$](/wp-content/uploads/2016/09/A339424_1_En_33_Chapter_Equ9.gif)
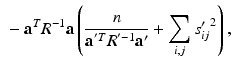
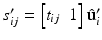 .
.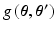 be the function obtained at the end of the E-step. We set the derivatives
be the function obtained at the end of the E-step. We set the derivatives  and
and 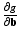 equal to 0 and solve for the parameters to obtain
equal to 0 and solve for the parameters to obtain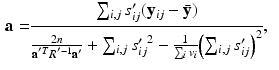
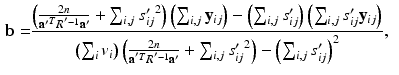
Related posts:
 Stable Overlapping Replicator Dynamics for Multimodal Brain Subnetwork Identification
Stable Overlapping Replicator Dynamics for Multimodal Brain Subnetwork Identification
 PET Reconstruction with Sparse Image Representation and Anatomical Priors
PET Reconstruction with Sparse Image Representation and Anatomical Priors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


