Fig. 5.1
Basic structure of normal lymph node. (a) Schematic representation of the anatomy of a lymph node with the cortical, paracortical, and medullary regions. The cortex contains multiple follicles including primary follicles and secondary follicles with germinal centers. (b) A secondary follicle with germinal center (GC) and well-defined mantle zone. In the GC, a loosely associated network of follicular dendritic cells, follicular helper T-cells, and centrocytes forms the light zone, whereas a dense area of dividing centroblasts can be recognized as a dark zone
Lymph Node Sampling and Tissue Handling
Excisional and core biopsies are generally most suitable for de novo evaluation of an enlarged lymph node. Fine needle aspiration cytology is often fraught with sampling limitations and does not permit evaluation of the lymph node architecture. In most clinical practices, lymph node samples are submitted either without fixative or in a nutrient-rich medium. Ideally, triage of tissue material from a patient with unexplained lymphadenopathy should be based on evaluation of a touch preparation. If neoplasia is a consideration, lymph node tissue should be handled in the following priority order: (1) formalin fixation for histologic evaluation (paraffin-embedded tissue may be also used for immunohistochemistry, fluorescence or colorimetric in situ hybridization, and molecular testing); (2) unfixed tissue in nutrient-rich media for immunophenotyping by flow cytometry; and (3) unfixed tissue in nutrient-rich media for cytogenetic analysis.
Overview of Diagnostic Imaging Approaches
Conventional Radiography/Fluoroscopy
Conventional radiographs (x-rays) are still commonly obtained as the initial imaging study in patients with disorders of the lymphoid system. Despite the emergence of sophisticated cross-sectional and functional imaging techniques, radiographs remain attractive for the initial evaluation, and they are inexpensive, rapid, and relatively easy to perform. They can be done without sedation and can be accomplished at far lower radiation doses than computed tomography (CT). Although many disorders will not be evident radiographically, the presence of primary intrathoracic tumors, mediastinal and/or paraspinal masses, intra-abdominal masses and destructive bone lesions can all be detected radiographically. The presence of calcifications in lymphoid masses may be helpful in establishing a differential diagnosis. For the evaluation of children with suspected abdominal conditions, radiographs of the abdomen should be the initial examination of choice to assess for acute obstruction or bowel perforation.
Historically, fluoroscopy was the primary modality used to evaluate the gastrointestinal tract and the upper airway. With the emergence of cross-sectional imaging, fluoroscopy is now rarely used for the primary evaluation of suspected lymphoid lesions. However, children with poorly characterized symptoms and physical exam findings may undergo fluoroscopic evaluation to assess the intestinal tract, and the radiologist should be aware of the imaging findings associated with lymphoid lesions involving the GI tract. Mucosa-based processes such as esophagitis, gastritis, or duodenitis are diagnosed almost exclusively by endoscopy nowadays. However, esophageal and intestinal strictures may benefit from further delineation by fluoroscopy, which can be performed safely and effectively. Current fluoroscopic techniques generally employ very low radiation doses due to advances in pulsed fluoroscopy, image intensifier sensitivity, and improved automatic dose rate controls [1]. Both water-soluble contrast or barium enteric contrast agents used in conjunction with fluoroscopy are considered safe even in immunocompromised children. While there are few contraindications to fluoroscopic evaluation of the intestinal tract, contrast enemas are generally avoided in neutropenic patients and barium enteric contrast should be avoided when bowel stricture or perforation are suspected.
Ultrasound
Ultrasound is an excellent technique for evaluating the child with a lymphoid system disease. With the availability of an array of high and low frequency transducers, ultrasound can be used to evaluate both superficial and deeper structures and is particularly useful in evaluating lymph nodes in the neck and axilla as well as the abdominal visceral organs [2]. Diagnostic ultrasound is relatively quick, inexpensive, and does not utilize ionizing radiation. In the hands of a skilled operator, ultrasound can also be used to evaluate even the most anxious child and does not usually require sedation. In contrast to adults, ultrasound images in children are usually of very high quality due to the relative paucity of intra-abdominal fat and other attenuating soft tissues seen in older patients and adults.
Color and pulse wave Doppler as well as the increasing availability of three-dimensional imaging techniques allow dynamic flow and vascularity to be assessed. This can be helpful in distinguishing between hyperemic/inflamed lymphoid tissue and normal tissue. The use of microbubble ultrasound contrast agents is still experimental and not routinely available for use in children [3]. The use of sonoelastography to measure the compressibility and elasticity of tissues—initially used in the evaluation of the liver—is presently under evaluation to determine tissue characteristics of superficial lymphoid structures in an effort to discriminate benign from neoplastic nodal enlargement [4].
Computed Tomography
Since its emergence in the 1970s, the use of CT has become routine in the evaluation of patients with suspected abnormalities of the lymphoid system. With the development of multiple detector row and ultrafast CT imaging techniques, examinations of the entire body can now be performed in under one minute and, depending on the area of interest, even young children may be evaluated without the need for sedation.
CT scanning is routinely performed in transaxial cross-sections with multiplanar reconstructions being rapidly generated from the axial image data. With the use of multidetector helical CT scanners and isotropic imaging acquisition techniques, the reconstructed images have no discernible loss of resolution or contrast as compared to source transaxial data. The use of contrast agents (both intravenous and enteric) in pediatric CT scanning depends on the condition being evaluated. For assessment of the mediastinum and lymphoid tissues, the presence of contrast is considered helpful, if not essential, to better characterize lymph nodes relative to adjacent vascular structures. In addition, the use of contrast-enhanced CT scanning remains the standard of care to accurately measure the size of lymphoid tumors to determine response to therapy [5, 6]. Although concerns regarding reactions to iodinated intravenous contrast agents are frequently raised, the rate of adverse reactions, including acute allergic or anaphylactoid reactions, asthmatic reactions, and vasovagal reactions, are exceedingly rare [7]. The presence of a prior contrast reaction does, however, predict subsequent recall-type allergic reactions, and clinicians and radiologists should be aware of the increased potential for adverse events in these patients and provide appropriate premedication as indicated. Awareness by radiologists and clinicians of guidelines for the use of iodinated contrast agents is important so that imaging protocols can be modified as needed [8].
Over the past 10 years, there has been a heightened awareness of the potential risk posed by CT scanning from ionizing radiation doses used [9]. When comparing the effective dose for a pediatric chest CT to a chest radiograph, some estimates indicate the CT scan is the equivalent of tens to hundreds of chest radiographs [10]. While it is clear that the use of multiple CT scanning phases and the need for multiple sequential CT examinations increases the relative radiation risk, a more extensive discussion of the radiation risk estimates associated with CT scanning in pediatric patients is beyond the scope of this chapter and can be reviewed in a number of recent publications [11–14]. It is reasonable to conclude that opinions vary widely and uncertainty about the absolute risk of cancers associated with ionizing radiation in the diagnostic CT dose range remains [15]. Nonetheless, the principle that guides the use of CT scanning in pediatric patients is adherence to the ALARA (as low as reasonably achievable) guidelines, which seek to balance the relative risk of exposure to ionizing radiation against the potential benefit from the diagnostic information obtained. Namely, every effort should be made to adjust the CT doses to the patient’s body size and clinical indications and to minimize scanning of body regions that are not involved or not suspected as harboring disease.
Most major manufacturers now have new dose-reducing automatic tube current modulation technologies as well as iterative image reconstruction algorithms that allow scans to be performed at much lower doses and for the doses to be adjusted dynamically, accounting for the differing tissue densities being imaged [16]. While the technical parameters used in CT scanning are ultimately the responsibility of the radiologist, clinicians ordering CT scans should be aware of these considerations in determining the need for CT and whether the clinical indications warrant the particular examination being ordered.
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI), in contrast to the transmission technologies such as conventional radiography, fluoroscopy, and CT, belongs to the group of emission technologies. MRI consists of dynamic interactions between a strong external magnetic field, the application of radiofrequency waves, and subsequent reception of low level radiofrequency (RF) emissions from hydrogen nuclei within the body [17]. Once the patient is placed in a magnetic field, the hydrogen nuclei become aligned in the orientation of the magnetic field. When they are bombarded with radiofrequency pulses, the hydrogen nuclei absorb the RF energy. Once the RF waves are turned off, the hydrogen nuclei begin to release the absorbed energy (“decay”) in a manner that is related to the tissue characteristics within which the nuclei are residing. This allows exquisitely detailed images of the body’s soft tissues to be acquired. There are a number of factors that can influence the signal and image quality obtained during MRI, but these are beyond the scope of this review. In general, in order to acquire a diagnostically useful image, contrast between normal tissues and diseased tissues is needed. Such contrast differences are an inherent reflection of the different tissue environments within which the protons live. Depending on the clinical indication, the MRI pulse sequences can be tailored to address specific clinical questions.
For evaluating the patient with abnormalities of the lymphoid system, characterization of normal and diseased lymph nodes can usually be accomplished well using standard T1- and T2-weighted imaging techniques in axial and at least one orthogonal (coronal or sagittal) imaging plane. The use of gadolinium-based contrast agents can be helpful in further characterizing tissue abnormalities. In some instances, the presence of iron or hemosiderin deposition may be helpful in rendering a differential diagnosis and gradient echo or T2-weighted images, which are sensitive to the local magnetic field inhomogeneities induced by the presence of paramagnetic compounds such as iron, can be utilized [18, 19].
Pediatric whole-body MRI techniques with rapid scanning sequences can now be accomplished typically in 30 min or less and have been advocated for staging and screening patients with abnormalities of the lymphoid system [20, 21]. The use of parallel imaging techniques and the ability to simultaneously receive RF information using multiple receiver coils allows for more rapid imaging and improved temporal and spatial resolution. The increased ability to perform rapid MRI studies for a wide variety of pediatric indications is particularly important for young children in whom an MRI examination of 30 min or more may require sedation in order to minimize patient motion.
The use of diffusion-weighted imaging has recently gained attention for body imaging techniques [22–24]. Diffusion-weighted imaging was initially developed for characterizing sites of brain ischemia in neuroradiologic applications. For oncologic purposes, the ability to evaluate tissue density and tumor cellularity based on changes in the restricted diffusion of water molecules within a particular region, has been helpful in characterizing and evaluating effects of therapy [24]. While diffusion-weighted imaging may aid in detecting small lymph nodes, it has not yet been able to reliably distinguish between neoplastic and non-neoplastic lymph node processes [25, 26].
The use of MR contrast agents includes both positive and negative contrast agents. Gadolinium (Gd)-based agents rely on the paramagnetic effects of Gd, which causes both T1 and T2 relaxation time shortening. The Gd ion itself is toxic and is only provided in the form of a gadolinium chelate. The pattern of tissue uptake and excretion may vary depending on the chelate used, but most commercially available Gd-based MR contrast agents are administered intravenously and are primarily excreted through the genitourinary tract. Iron- and iron oxide-based contrast agents have been specifically advocated for evaluation of the lymphoid system [19]. These agents rely on the T2 shortening effects of iron. Normal cells of the macrophage and reticuloendothelial cell lineage will typically scavenge the iron-based nanoparticles, and the use of these contrast agents has been shown in several pilot studies to be helpful in distinguishing lymph nodes involved by tumor from normal lymph nodes. Despite the potential appeal of this class of contrast agents, they have not received universal clinical acceptance.
The use of Gd-based chelates is relatively safe, although patients with underlying acute or chronic renal insufficiency have been shown to be at risk of developing nephrogenic systemic fibrosis (NSF) [27]. NSF is characterized by progressive tissue fibrosis that may be limited. On occasion, NSF may be progressive with involvement of other tissues such as the heart, lungs, and esophagus resulting in significant pathology and potentially death. Although the risk of developing NSF seems to relate to the doses of Gd chelate used and the degree of renal insufficiency, the majority of patients with renal insufficiency do not develop NSF. Nonetheless, the evaluation of renal function prior to the intended use of Gd-based contrast agents is needed in order to minimize this potential risk.
Nuclear Medicine/Positron Emission Tomography
Functional imaging techniques in nuclear medicine are commonly used in evaluating patients with abnormalities of the lymphoid system. Although these techniques provide less anatomic and morphologic information than either CT or MRI, there is often valuable metabolic and functional information to be gathered from specific nuclear medicine techniques. As with MRI, nuclear medicine imaging relies on emission data following injection with a radiopharmaceutical agent to generate an image [28]. Depending on the pharmaceutical and isotope utilized, the agent localizes to target organs and tissues and is visualized using highly specialized detector cameras used to acquire and display images of the whole body or selected body regions. Cameras are configured to acquire both 2-D planar as well as multiplanar tomographic images and can be placed at various angles and positions to optimize image quality and lesion conspicuity.
Positron emission tomography (PET) utilizes positron-emitting radiopharmaceuticals. Following radionuclide decay in situ, emitted positrons annihilate upon contact with electrons near the point of tissue localization releasing two 511 keV photons traveling in opposite directions. These high-energy photons can be imaged using coincidence detection techniques to permit precise identification of the initial site of radiopharmaceutical localization and positron decay. Similarly, single photon emission computed tomography (SPECT) applies tomographic rather than planar imaging techniques to gamma particle emitting radiopharmaceuticals allowing multiplanar acquisition and projection images to be generated. These can be fused to CT and/or MRI images, which in some instances may be simultaneously acquired to allow anatomical and functional co-localization.
A wide variety of radiopharmaceuticals is available, although the main agents used in routine evaluation of children with lymphoid disorders include 99mTc labeled methylene diphosphonate (MDP), 67Ga, and 18F-fluorodeoxyglucose (FDG). 99mTc-MDP localizes to areas of bone turnover and is useful in assessing osteoblastic metastatic disease as well as sites of lytic bony disease. Historically, 67Ga, whic binds to transferrin receptors expressed on lymphoid cells, was used to evaluate patients with lymphoma. The use of gallium scanning has been essentially replaced by FDG PET imaging, and the latter has become standard of care in the evaluation of patients with lymphoma [29, 30] as well as a variety of other non-neoplastic lymphoid disorders [31]. FDG PET imaging relies on the presence of 18F-fluorine as a positron-emitting radiotracer that is incorporated into the glucose analog fluorodeoxyglucose. FDG accumulates at relatively high levels in metabolically active lesions including lymphoid malignancies and inflammatory disorders, providing a very sensitive means of evaluating patients with abnormalities of the lymphoid system. Nearly all PET imaging is currently performed with integrated PET/CT scanners. This allows areas of FDG uptake at sites of concern to be fused to simultaneously acquired CT images to afford better anatomic correlation and coregistration of uptake sites. In addition, areas of background or physiologic uptake such as brown fat can be accurately identified using co-registered PET/CT images.
Other nuclear medicine imaging techniques include the use of 99mTc-HMPAO or 111In-oxine labeled leukocytes for inflammatory imaging, although this technique is now less frequently used with the advent of FDG PET imaging. Nuclear medicine plays an important complimentary role in providing a functional characterization of patients with disorders of the lymphoid system. In many diseases, functional imaging is crucial for disease staging, response assessment, and in certain instances disease surveillance. Increasingly, however, the use of nuclear medicine techniques is closely integrated with simultaneously acquired cross-sectional imaging examinations (e.g., CT or MRI), requiring careful assessment of both the functional and anatomic imaging data for diagnostic evaluation [29, 30, 32].
Benign Causes of Lymphadenopathy
In clinical practice, infectious and reactive etiologies are nearly always in the differential diagnosis of lymphadenopathy in a young patient [33]. The most common sites for lymphadenitis requiring imaging evaluation are the cervical, axillary, and inguinal regions. Clinically, lymphadenitis is associated with pain, tenderness, and focal lymph node enlargement [34, 35]. The most common causes of infectious lymphadenitis are summarized in Table 5.1.
Table 5.1
Most common infectious lymphadenitis
Etiology | Common anatomic site | Histology | Diagnosis |
|---|---|---|---|
Viral | |||
Epstein-Barr virus (EBV)-infectious mononucleosis | Cervical lymphadenopathy and splenomegaly | Interfollicular infiltrate with numerous immunoblasts (EBV positive) | Monospot test, and serology studies |
Cytomegalovirus | Localized or generalized lymphadenopathy | Florid follicular hyperplasia and hyperplasia of monocytoid B-cells | Presence of CMV inclusions on infected cultured cells |
Herpes simplex virus | Localized or multifocal lymphadenopathy | Interfollicular hyperplasia with necrotic areas with neutrophils and karyorrhectic debris | Presence of cells with glass-like inclusions in the areas of necrosis |
Bacterial | |||
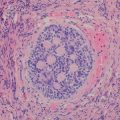
Bartonella henselae (Cat-scratch disease) | Regional lymphadenopathy following a skin lesion | Stellate necrotizing granulomas with palisading epithelioid histiocytes | Warthin-Starry stain, PCR, and serology |
Common bacteria (streptococcal or staphylococcal) | Cervical lymphadenopathy | Follicular hyperplasia, and microabscesses in the lymph node | Aerobic and anaerobic tissue cultures |
Mycobacterium tuberculosis | Supraclavicular and cervical lymph nodes | Ceseating granulomas with epitheliod histiocytes and multinucleated giant cells | Ziehl-Neelsen stain, cultures and PCR |
Nontuberculous Mycobacteria (MAI, M. kansasii, M. scofulaceum, M. malmoense) | Unilateral cervical lymph nodes | Ceseating granulomas with epitheliod histiocytes and multinucleated giant cells | Ziehl-Neelsen stain, PAS + MAI and PCR |
Protozoal | |||
Toxoplasma | Unilateral or bilateral lymph nodes | Florid follicular hyperplasia, monocytoid B-cells and clusters of epithelioid histiocytes surrounding germinal center | Characteristic cervical histologic changes and serology |
Various infectious and reactive etiologies are often associated with broad histologic patterns of lymphadenopathy that include follicular, paracortical, sinusoidal, and mixed patterns [36]. While a more detailed discussion of the pathophysiologic causes that underlie these patterns is beyond the scope of this textbook, a summary of the most common etiologies associated with the various histologic patterns is listed in Table 5.2. Progressive transformation of germinal centers (PTGC) is a term used to describe a well-defined histologic pattern of follicular changes often associated with reactive lymphadenopathy. PTGC is characterized by follicles of varying sizes exhibiting disruption of the germinal center by encroaching mantle zone lymphocytes. The etiology and pathogenesis of isolated PTGC remain poorly understood. While the vast majority of PTGC changes are seen in benign reactive lymph nodes, recognition of this change is important since it is also often seen in the vicinity of nodular lymphocyte predominant Hodgkin lymphoma.
Table 5.2
Common reactive lymphadenopathies
Pattern | Histologic findings | Entity |
|---|---|---|
Follicular | Numerous reactive follicles; GC with irregular shape and size Tingible body macrophages; BCL2 is negative in GC | Follicular hyperplasia Autoimmune disorders Castlemans disease, hyaline vascular type Progressive transformation of germinal centers |
Sinusoidal | Prominent expansion of sinuses Histiocytic hyperplasia Plasma cell proliferation Monocytoid B-cells | Sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease) Storage disease Whipple disease Vascular transformation of sinuses Hemophagocytic syndrome |
Interfollicular mixed | Interfollicular expansion T-cell predominance Immunoblasts increased in the interfollicular areas Plasma cells, macrophages Mixed population | Dermatopathic lymphadenopathy Kimura’s disease Systemic lupus erythematous Kicuchi’s lymphadenitis Inflammatory pseudotumor |
Diffuse | Effacement of architecture Mixed population Immunoblastic proliferation | Drug-induced lymphadenophathy Viral lymphadenitis |
Lymphoproliferative Disorders and Neoplasms Associated with Immune Deficiency
Lymphoproliferative Disorders and Neoplasms Associated with Primary Immunodeficiency Diseases
Primary immunodeficiencies (PID) are a rare and heterogeneous group of diseases caused by congenital defects affecting the innate and/or adaptive immunity, with impact on the humoral and/or cell-mediated immunity. The most updated classification for PID provided by the International Union of Immunodeficiencies Studies recognizes eight categories [37]: (1) combined immunodeficiencies: e.g., severe common immunodeficiency (SCID); (2) predominantly antibody deficiencies: e.g., X-linked agammaglobulinemia (XLA), common variable immunodeficiency (CVID); (3) well-defined syndromes with immunodeficiency: e.g., Wiskott-Aldrich syndrome, DNA repair defects; (4) diseases of immune dysregulation: e.g., autoimmune lymphoproliferative syndrome (ALPS), familial hemophagocytic lymphohistiocytosis; (5) congenital defects of phagocyte number, function or both: e.g., chronic granulomatous disease, severe congenital neutropenia; (6) defects in innate immunity: e.g., deficiencies impairing the interferon gamma/interleukin 12 axis; (7) autoinflammatory disorders: e.g., familial Mediterranean fever; and (8) complement deficiencies. More than 120 distinct genes have been identified, with abnormalities that account for more than 150 different forms of PID [38].
Patients with PID have a higher than usual risk of developing lymphoid malignancies, and these represent the second leading cause of death in this group [39]. The incidence of lymphoproliferative disease ranges from 1.4 to 24 % depending of the type of the PID. The diseases commonly associated with lymphoproliferative disorders or neoplasms include ataxia-telangiectasia, Wiscott-Aldrich syndrome, Nijmegen breakage syndrome, CVID, XLA, and autoimmune lymphoproliferative syndrome. In these patients, lymphoma is diagnosed at a median age of 7.1 years [39].
The pathogenesis of lymphoproliferative diseases in PIDs involves multiple mechanisms rooted in defects in DNA damage response (e.g., ataxia-telangiectasia and Wiscott-Aldrich syndrome) and immune deregulation that characteristically results in abnormal response to viral infections such as Epstein-Barr virus (EBV), hepatitis B or C viruses, and human papillomavirus [40]. The inability to eliminate infection is speculated to create an inflammatory environment that can eventually promote tumor development and growth by promoting and sustaining the acquisition of oncogenic somatic mutations [39].
Lymphoid proliferations associated with PID are heterogeneous and range in spectrum from reactive hyperplasia to non-Hodgkin and Hodgkin lymphoma. The most common presentation is at extranodal sites; namely, the gastrointestinal tract, lungs, and central nervous system. In patients with XLA and SCID, non-neoplastic lesions include a polymorphous lymphoid proliferation with plasmablasts, immunoblasts and Reed-Sternberg-like cells that result from primary EBV infection. Patients with CVID can present with lymphoid hyperplasia in the gastrointestinal tract or with lymph node follicular hyperplasia and expansion of paracortical areas by immunoblasts infected by EBV. In patients with ALPS, lymphadenopathy results from follicular hyperplasia with or without progressive transformation of germinal centers. Overt lymphomas generally have clonal IgH rearrangement whereas non-neoplastic or polymorphous lymphocytic proliferations may show either oligoclonal or monoclonal IgH rearrangements. The presence of clonality in patients with PID does not always indicate the presence of lymphoma and correlation with clinical and imaging findings is particularly important.
The lymphomas that can occur in patients with PID are generally classified like those that arise in non-immunosuppressed patients. Diffuse large B-cell lymphoma is the most common type, followed by Burkitt lymphoma. Lymphomatoid granulomatosis involving the lungs shows T-cell background infiltrate and neoplastic B-cells, and is most commonly seen in patients with Wiscott-Aldrich syndrome. Classical Hodgkin lymphoma is less common than diffuse large B-cell lymphoma in PID patients and has been reported in patients with ataxia-telangiectasia syndrome and Wiscott-Aldrich syndrome. Peripheral T-cell lymphoma is rare and has been described in patients with ALPS. The EBV virus has been demonstrated to be present in most of the cases [41]. A summary of the most common PID types associated with lymphoproliferative disorders and neoplasms is listed in Table 5.3.
Table 5.3
Most common primary immunodeficiencies associated with lymphoproliferative disorders
Type of deficiency | Gene defect | Pathogenesis | Abnormality | Lymphoproliferative disease |
|---|---|---|---|---|
CVID, autosomal (predominantly antibody deficiency) | ICOS, CD19, BAFFR | Defects in B-cell maturation and differentiation | Reduced B cells; low IgG and IgA | GI tract and lungs (HHV8 implicated in granulomatous interstitial lung disease), increased risk of B cell, extranodal NHL |
SCID (combined immunodeficiency) | X-linked, γ chain mutations; JAK3, AR; ADA, AR | Abnormal interleukin signaling in XSCID and JAK3 mutations; accumulation of dATP in defective ADA | Low or absent T-cells and NK cells and nonfunctional B cells | Fulminant primary EBV Infection, lymphadenopathy, hepatosplenomegaly. Lymphoma development not well documented |
Hyper IgM syndrome, X-linked (predominantly antibody deficiency) | CD40/CD40L ligand mutation | Defective B cell and dendritic cell signaling; CD40L required for isotype class-switching from IgM to IgG or IgA | Low or absent IgG, IgA; normal or increased IgM, cytopenias | Predominantly abdominal adenopathy; lack germinal of centers. Lymphoma development not well documented |
Ataxia-telangiectasis, AR (well-defined syndromes with immunodeficiency) | AT gene mutation (11q22–23), encodes for protein kinase | Defective DNA repair and deregulation of cell division and apoptosis, abnormal function and development of lymphocytes | IgA, IgE, IgG2 decreased; progressive decrease in T-cells; normal B cell numbers | T-cell acute lymphoblastic leukemia/lymphoma in children; T-prolymphocytic leukemia in young adults; Hodgkin lymphoma-like lymphoproliferative disorders |
X-linked lymphoproliferative syndrome (well-defined syndromes with immunodeficiency) | SH2D1A mutations (Xq25), that encodes for SAP protein | Uncontrolled T-cell proliferation; underlying mechanisms is not yet entirely understood | B cells normal or reduced. Defective lysis and polarization following EBV infection | Fulminant IM; extranodal B cell NHL, particularly of the terminal ileum |
Autoimmune lymphoproliferative syndrome (well-defined syndromes with immunodeficiency) | Defects in the Fas/FasL signaling | Defects in apoptotic pathway | Autoimmune cytopenias, adenopathy and splenomegaly | Increased risk for HL, including NLPHL |
Wiskott-Aldrich, AR (well-defined syndromes associated with immunodeficiency) | WAS mutation (Xp11.22–23) | Abnormal cytoskeletal architecture of hematopoietic cells | Decrease in T-cells, normal B cells; decreased IgM | 7 % develop NHL; CNS involvement has been reported in this setting; Lymphomatoid granulomatosis |
Imaging Features
Imaging features associated with PID vary by the type and severity of the immunodeficiency. Patients with XLA do not produce immunoglobulin but do have normal T-cell function and normal thymic development. Radiographically, this disorder is most frequently associated with the sequelae of recurrent pulmonary infections such as bronchiectasis, pulmonary parenchymal scarring and heterogeneous or mosaic patterns of lung aeration and attenuation (Fig. 5.2). Bacterial infection can also occur at other sites such as the sinuses and genitourinary tract; however, these are not as commonly encountered as pulmonary infections [42]. Patients with IgA deficiency develop pulmonary and GI infections and may have underlying allergic and autoimmune disorders. In comparison to XLA, the imaging findings are less severe and bronchiectasis, for example, is less common. Patients with CVID, as with XLA, may also have recurrent infections, bronchiectasis, and bronchial wall thickening. In contrast to the other PID, patients with CVID may also develop lymphadenopathy and splenomegaly in addition to a more generalized lymphoproliferative process (Fig. 5.3), presumably because of varying degrees of associated T-cell functional abnormalities [42, 43].
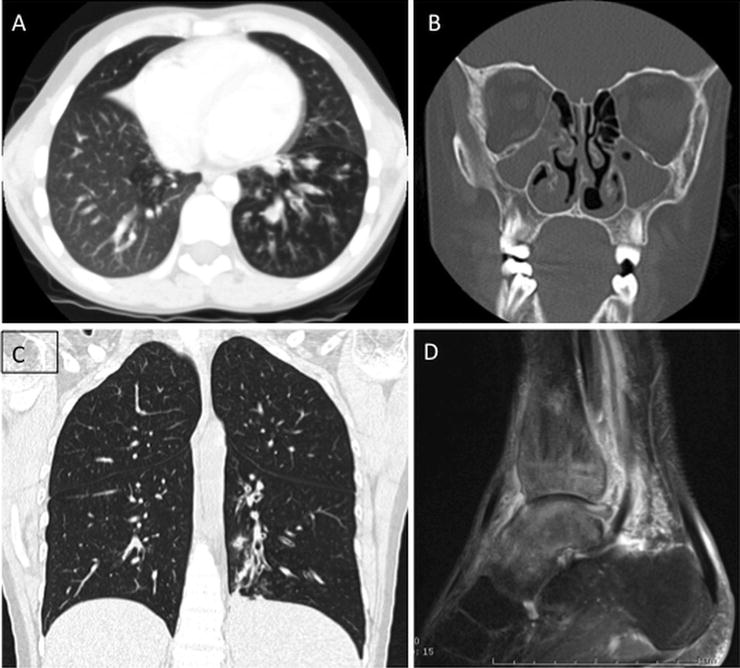
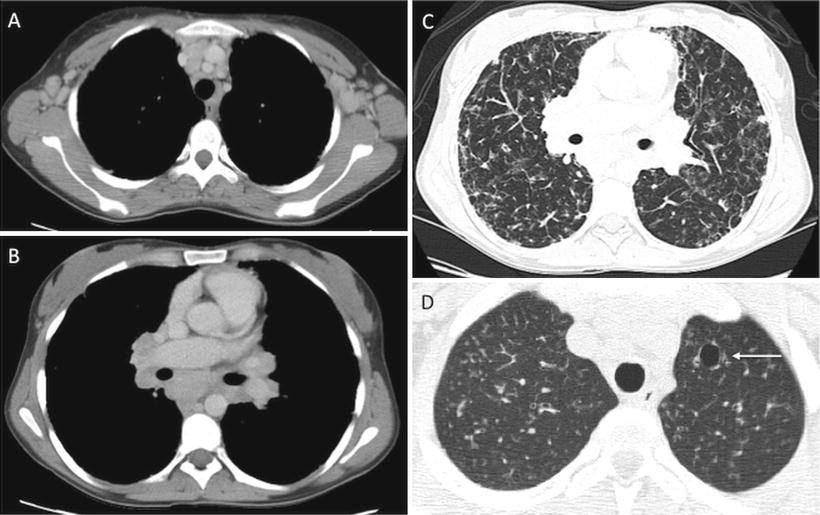
Fig. 5.2
X-linked agammaglobulinemia. Three examples of findings in patients with XLA. 9-year-old with chronic cough and sinus infection with chest CT showing mosaic lung attenuation and air trapping in the left lower lobe (a) and near complete opacification of the maxillary sinuses (b). Chest CT in a 32-year-old patient with XLA and respiratory symptoms shows left lower lobe bronchial wall thickening, bronchiectasis, and evolving consolidation (c). Ankle MRI (d) in a 30-year-old patient with XLA revealed osteomyelitis and tenosynovitis related to chronic infection
Fig. 5.3
Common Variable Immunodeficiency (CVID) (a–c) and Hyper-IgE syndrome (d). Chest CT in a 26-year-old patient with CVID (a–c) shows axillary (a) and mediastinal/hilar lymphadenopathy (b), in addition to extensive parenchymal lung disease (c), with ground glass opacities, reticular thickening and areas of air trapping related CVID. Chest CT in patient with hyper-IgE syndrome reveals a small pneumatocele (arrow), characteristic of this disease, in the left upper lobe, as well as background diffuse nodular opacities consistent with underlying infection
DiGeorge syndrome (DGS) and SCID are the most severe of the PID syndromes. DiGeorge syndrome, also known as thymic hypoplasia, results in primary T-cell deficiency secondary to abnormal thymic development [42]. Patients with DGS have other findings including hypertelorism and micrognathia, which may be apparent radiologically. Other associated anomalies include a host of cardiovascular anomalies such as abnormalities of the aortic arch and tetralogy of Fallot. Apart from the narrow mediastinum resulting from absent thymic tissue, there are no other specific imaging findings to indicate the diagnosis of DGS. Severe combined immunodeficiency syndrome results from absent T- and B-cell function, as well as diminished natural killer cell function. As with DGS, the combination of deficiencies in both humoral and cellular immunity in patients with SCID leads to recurrent and severe opportunistic infections [43].
Patients with Wiskott-Aldrich syndrome and ataxia telangiectasia have a high incidence of developing malignancy, particularly leukemia and lymphoma [44, 45] (Fig. 5.4). In addition, patients with ataxia telangiectasia are at an increased risk of radiation-induced malignancy, which creates unique challenges for surveillance imaging studies. Because imaging examinations in these populations of patients are aimed at screening for malignancy or characterizing active or chronic sites of infection, the results are often nonspecific, prompting multiple follow-up imaging evaluations. As such, techniques that require repeated exposure to ionizing radiation (i.e., CT) should be used judiciously [46].
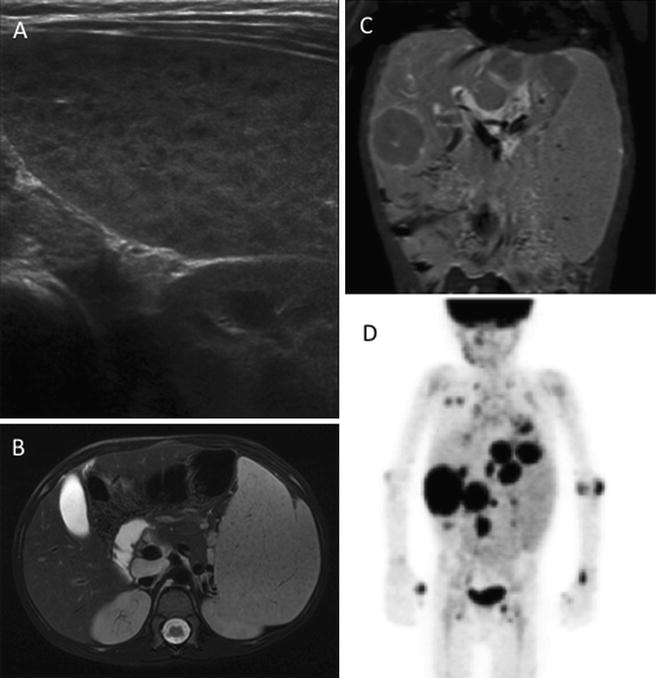
Fig. 5.4
A 6-year old with ataxia telangiectasia initially presented with splenomegaly. Ultrasound showed diffuse splenic enlargement with innumerable hypoechoic lesions, concerning for lymphoproliferative disease (a). Over the next 2 months subsequent MRI confirmed splenic involvement (b), in addition to showing multifocal hepatic involvement (c). Biopsy revealed large cell lymphoma. FDG PET shows extensive abnormality of the abdomen, lungs, and skeleton (d)
Lymphoid Neoplasms Associated with Human Immunodeficiency Virus Infection
Human immunodeficiency virus (HIV) infection and the associated acquired immune deficiency syndrome (AIDS) occur infrequently in children and often as the result of in utero transmission. A distinct group of lymphoid neoplasms arise in the setting of HIV infection. These include lymphomas that are seen in HIV-negative patients such as Burkitt lymphoma, diffuse large B-cell lymphoma, and Hodgkin lymphoma. In addition, HIV patients are affected by lymphomas that are only rarely seen outside the setting of HIV: primary effusion lymphoma and plasmablastic lymphoma. The introduction of highly active antiretroviral therapy (HAART) has resulted in a dramatic decrease in the incidence of high grade lymphomas in HIV patients.
Most patients present with advanced stage disease and extranodal involvement. Extranodal sites usually include the gastrointestinal tract, central nervous system (CNS), liver and bone marrow, as well as more unusual sites such as oral mucosa and body cavities. Lymph nodes can also be involved. Epstein-Barr virus is identified in approximately 40 % of HIV-associated lymphomas, but this percentage varies significantly between the different types of lymphomas. Distinction between neoplasia and infection on imaging studies may be difficult occasionally; for example, CNS involvement by toxoplasmosis is a rare complication in pediatric HIV patients and can be difficult to distinguish from lymphoma [47].
Post-transplant Lymphoproliferative Disorders
Post-transplant lymphoproliferative disorders (PTLD) are lymphoid or plasma cell disorders that develop in a recipient of a solid organ or allogeneic stem cell transplant as a result of iatrogenic immunosuppression. The spectrum of PTLD is clinically and histologically variable. Three broad categories of PTLD are recognized: (1) early lesions (infectious mononucleosis-like and plasmacytic hyperplasia); (2) polymorphic PTLD; and (3) monomorphic PTLD. The latter includes lymphomas that also occur in non-immunosuppressed patients such as Hodgkin lymphoma and plasma cell myeloma. When compared to the adult population, the incidence of PTLD in children is higher, especially following solid organ transplant. The risk for development of PTLD is significantly associated with the occurrence of primary EBV infection after transplantation [48, 49].
Imaging of PTLD patients is directed at establishing the extent of involvement and should include cross-sectional imaging, either CT or MRI, of the entire torso as well as FDG PET imaging to establish the overall extent of disease [50]. Both CT and PET can be used to monitor response to therapy and/or immunomodulation. FDG PET has been shown to be superior to conventional imaging techniques in the early evaluation of PTLD response to therapy [50].
Routine screening for PTLD (e.g., EBV serologic studies or quantitative assessment of viral RNA) facilitates early diagnosis and treatment by reduction of immunosuppression. Lymph nodes as well as extranodal sites (tonsil, gastrointestinal tract, lungs, spleen and liver can be involved (Fig. 5.5). In solid organ transplant recipients, PTLD may affect the allograft itself and needs to be differentiated from rejection or infection. Detection of EBV-encoded RNA in the lesion generally favors PTLD. Early lesions, including infectious mononucleosis-like PTLD are more frequent in children and usually involve tonsils and adenoids. They have a good prognosis and respond to reduction of immunosuppressive treatment. However, the polymorphous PTLD is the most common type affecting pediatric transplant recipients. Some cases of polymorphic PTLD and most cases of monomorphic PTLD require additional treatment that includes monoclonal antibodies against B-cell antigens (e.g., rituximab), chemotherapy, and more recently cellular immunotherapy (EBV-specific cytotoxic T-cell immunity).
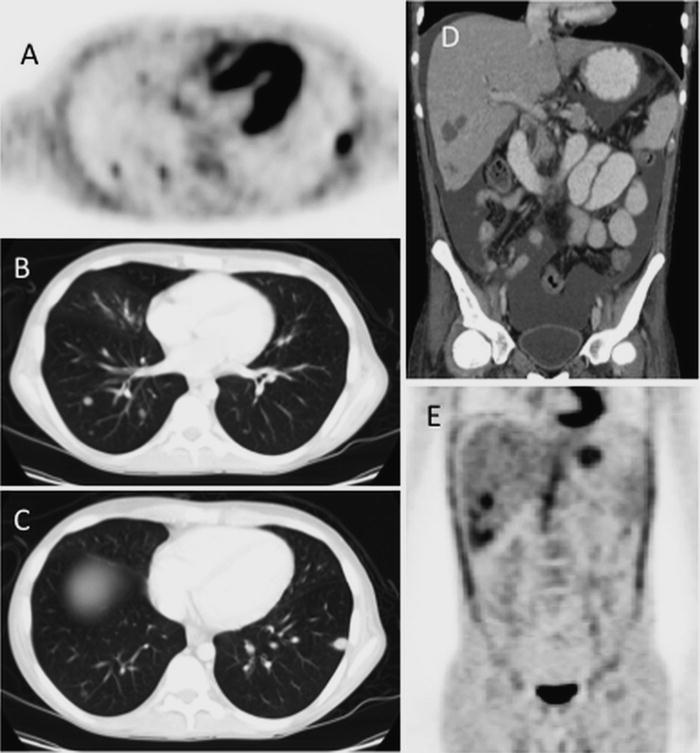
Fig. 5.5
PTLD involving lung and liver: 16-year old with AML s/p BMT with development of pulmonary PTLD. FDG-PET (a) and Chest CT (b, c) show multiple FDG avid lung lesions. Abdominal CT (d) and accompanying FDG-PET (e) show multiple hepatic lesion as well as ascites, all consistent with multi-focal sites of PTLD involvement
Overview and Classification of Lymphoid Tumors
Lymphoma represents the third most common pediatric malignancy [51]. Lymphoid malignancies comprise 11 % of total cancers diagnosed in children and 13 % in adolescents and young adults (15–39 years) [52]. The classification of lymphoid neoplasms has undergone a bewildering series of changes over the course of several decades. Those classifications have been universally supplanted by the World Health Organization (WHO) classification. Distinct entities in the WHO classification are defined on the basis of morphologic, immunophenotypic, molecular, and clinical features. Lymphoid neoplasms in the current WHO classification that generally affect children and adolescents are summarized in Table 5.4.
Table 5.4
Common lymphoid neoplasms in children and young adolescents
Precursor lymphoid neoplasms |
B-lymphoblastic leukemia/lymphoma, NOS |
B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities |
T-lymphoblastic leukemia/lymphoma, NOS |
Mature B-cell neoplasms |
Nodal marginal zone lymphoma |
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) |
Pediatric follicular lymphoma |
Diffuse large B-cell lymphoma, NOS |
Lymphomatoid granulomatosis |
Primary mediastinal (thymic) large B-cell lymphoma |
ALK positive large B-cell lymphoma |
Burkitt lymphoma |
Mature T-cell neoplasms |
Epstein-Barr virus (EBV) positive T-cell lymphoproliferative disease of childhood |
Systemic EBV + T-cell lymphoproliferative disease of childhood |
Hydroa vacciniform-like lymphoma |
Extranodal NK/T-cell lymphoma, nasal type |
Enteropathy associated T-cell lymphoma |
Hepatosplenic T-cell lymphoma |
Subcutaneous panniculitis-like T-cell lymphoma |
Primary cutaneous gamma-delta T-cell lymphoma |
Peripheral T-cell lymphoma, NOS |
Anaplastic large cell lymphoma |
ALK positive |
ALK negative |
Hodgkin lymphoma |
Nodular lymphocytic predominant Hodgkin lymphoma |
Classical Hodgkin lymphoma |
Nodular sclerosis |
Mixed cellularity |
Lymphocyte-rich |
Lymphocyte-depleted |
Immunodeficiency –associated lymphoproliferative disorders |
Lymphoproliferative diseases associated with primary immunodeficiency |
Lymphomas associated with HIV infection |
Post-transplant lymphoproliferative disorders |
Plasmacytic hyperplasia and infectious-mononucleosis-like PTLD |
Polymorphic PTLD |
Monomorphic PTLD |
Classical Hodgkin lymphoma PTLD |
Histiocytic neoplasms |
Langerhans cell histiocytosis |
Disseminated Juvenile xanthogranuloma |
Precursor T and B Neoplasms
Together, precursor T and B neoplasms represent the most common malignancies in childhood. These neoplasms are derived from precursor lymphoid cells (lymphoblasts) arrested at an early stage of differentiation. B lymphoblastic leukemia/lymphoma accounts for approximately 85 % of cases while T lymphoblastic leukemia/lymphoma accounts for the remaining 15 % [53].
Lymphoid precursor neoplasms may either involve the bone marrow and blood (lymphoblastic leukemia) or exhibit a predominantly tissue-based distribution (lymphoblastic lymphoma). An arbitrary cutoff of ≥25 % blasts in the blood and/or bone marrow is used to define lymphoblastic leukemia. Since such a distinction has no known diagnostic or prognostic relevance, the current WHO classification employs the term lymphoblastic leukemia/lymphoma.
The prognosis of patients with precursor T and B neoplasms is based on many parameters that include molecular and cytogenetic features in addition to response to induction chemotherapy. In general, patients with T lymphoblastic leukemia/lymphoma tend to have worse outcomes than those with B lymphoblastic leukemia/lymphoma.
T Lymphoblastic Leukemia/Lymphoma
Clinical Features and Epidemiology
T lymphoblastic leukemia/lymphoma is a neoplasm of lymphoid progenitors committed to the T lineage. The disease commonly affects males in the second decade of life, and whites are more commonly affected than Asians or African Americans [54]. Patients with ataxia-telangiectasia have an increased predisposition to developing T lymphoblastic leukemia/lymphoma [45]. Up to 85 % of children and adolescents with T lymphoblastic leukemia/lymphoma present with a mediastinal mass and/or lymphadenopathy without leukemia [55, 56]. The majority of patients have symptoms of mediastinal involvement, including pain, facial swelling, and respiratory compromise. Depending on the size of the mediastinal mass, there may be a significant compression of the airway, cardiac, and central vascular structures.
Imaging Features
Patients with T lymphoblastic leukemia/lymphoma and a large mediastinal mass often have their initial disease suspected based on chest radiograph (Fig. 5.6a, b). These rapidly proliferating tumors can enlarge quickly and present one of the few oncologic emergencies one may encounter in the imaging suite [57]. The presence of a pericardial effusion may result in cardiac tamponade. Care should be taken with supine positioning of these patients and with sedation given the increased risk of respiratory compromise and impaired venous return.
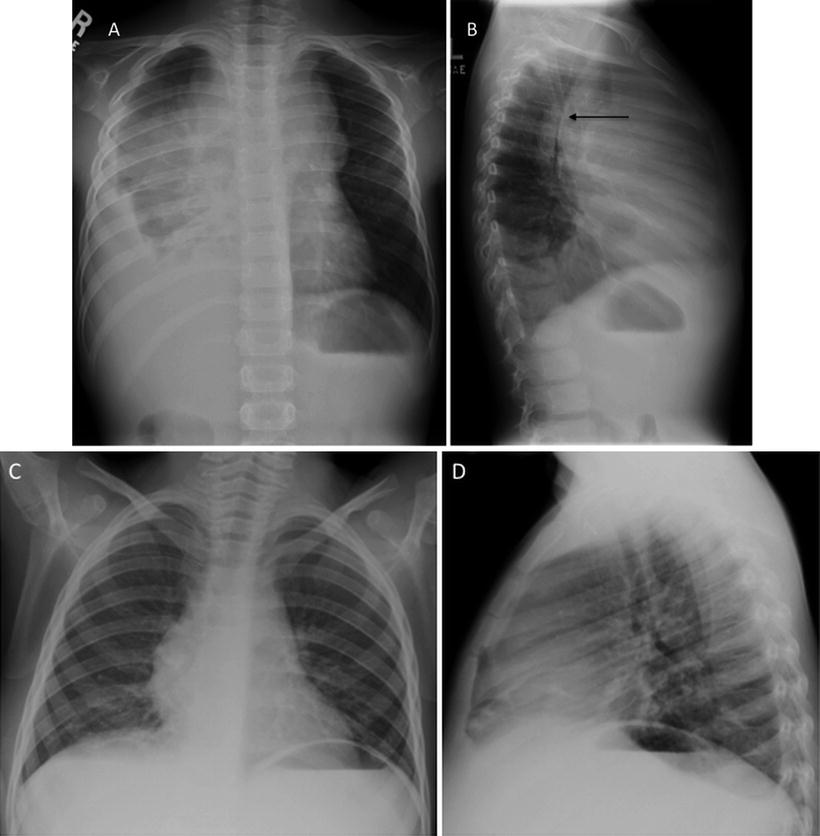
Fig. 5.6
T-lymphoblastic lymphoma: 5-year old with wheezing, fatigue, and pallor. AP and lateral CXR at diagnosis shows large mediastinal mass with significant tracheal narrowing evident on the lateral projection arrows and a large right pleural effusion (a, b). The patient could not lie flat for biopsy; pleural fluid aspirate showed T-Cell ALL. After just 6 days of therapy mediastinal mass has nearly resolved and effusions are gone (c, d)
Once appropriate therapy is instituted, patients with Tlymphoblastic leukemia/lymphoma typically have a rapid shrinkage in their mediastinal mass and improvement in their symptoms (Fig. 5.6c, d). There is no clear prognostic role for CT characterization of the mediastinal mass apart from establishing the extent of disease and sites of disease of involvement that may require urgent intervention. CT scans at the time of diagnosis may demonstrate extensive abdominal visceral involvement and intra-abdominal lymphadenopathy (Fig. 5.7). Some studies have shown that early resolution of the mediastinal mass, either by chest radiograph [58] or by CT [59] is associated with improved outcome, presumably reflecting patients with more chemosensitive disease.
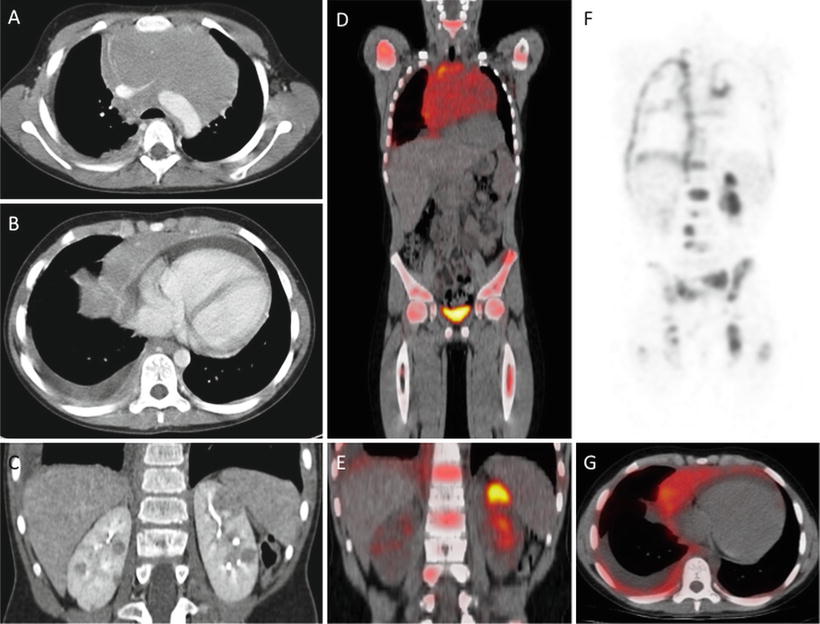
Fig. 5.7
T-lymphoblastic lymphoma, CT and FDG PET: 10-year old with T-ALL, mediastinal mass compressing major vessels (a), pleural effusion and pericardial effusion (b) and extensive visceral (bilateral renal (c) and bony disease (e, f). FDG PET/CT confirms multifocal sites of involvement by metabolically active tumor and in particular shows the extent of bone involvement (d–g)
Although these rapidly dividing tumors are quite metabolically active and typically FDG avid (Fig. 5.7), staging and post-treatment evaluation by FDG PET imaging is not used frequently in patients with precursor lymphoid neoplasms.
Pathology
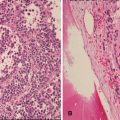
The neoplastic cells have scant to moderate cytoplasm, irregular nuclear contours, and a dispersed chromatin pattern. The latter is best appreciated on Wright-Giemsa-stained preparations. A similar pattern is noted on hematoxylin-and-eosin-stained tissue sections. Characteristically, the neoplastic cells have high rates of mitosis and apoptosis with frequent areas of increased background macrophages imparting a so-called starry-sky pattern (Fig. 5.8).
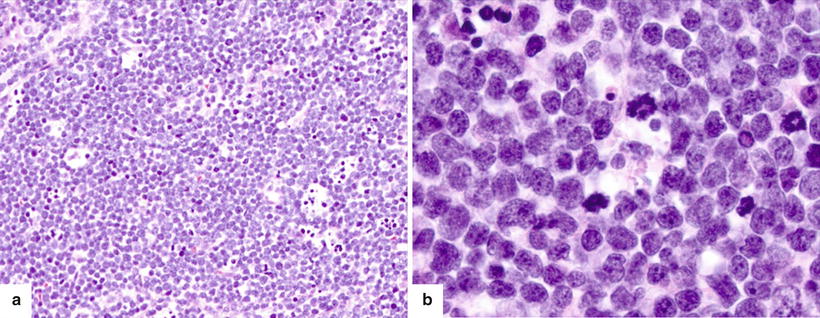
Fig. 5.8
(a) Precursor T-cell ALL diffusely involving the lymph node with numerous mitoses and tingible-body macrophages imparting a “starry-sky” appearance. (b) The blasts have characteristic features including small to intermediate size, scant amount of cytoplasm, irregular nuclear membranes, finely dispersed chromatin, and inconspicuous nucleoli
Immunohistochemistry or flow cytometry immunophenotyping demonstrates expression of one or more T lineage markers including CD1a, CD2, CD3, CD4, CD7 and CD8 (Fig. 5.9). Of these, CD3 is regarded as the bona fide T lineage-specific marker. In addition, the neoplastic cells are often positive for CD34, TdT, and CD99. It should be noted that CD99 expression may be positive in other small round cell tumors that are usually part of the differential diagnosis [60]. T-LBL can be further subclassified into groups representing stages of T-cell maturation and differentiation (Table 5.5). Early T-cell precursor (ETP) acute lymphoblastic leukemia/lymphoma represents approximately 15 % of precursor T neoplasms and is characterized by high frequency of relapses following standard chemotherapy [61]. This recently described entity is characterized by CD3 expression (cytoplasmic ± surface using flow cytometry), lack of expression of CD1a, and aberrant expression of stem cell myeloid markers (CD34, CD33, CD13, and CD117).
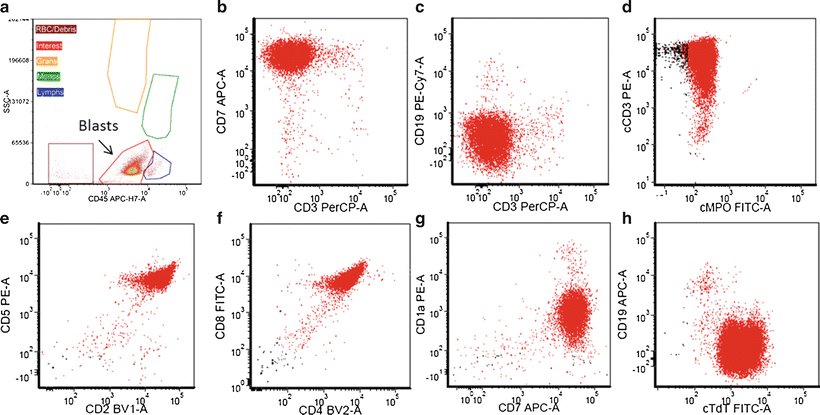
Fig. 5.9
Precursor T-cell ALL immunopheotype. The patient is a 14-year-old male with diffuse lymphadenopathy. A core biopsy reveals a diffuse infiltrate of mononuclear cells with blast morphology. Flow cytometry was performed on a cell suspension that showed increased blasts defined by dim CD45 expression and low side scatter (SSC-A) properties (a). The blasts expressed bright CD7 (b), and they were negative for CD19 (c). They also expressed cytoplasmic CD3 that confirmed T-cell lineage, while they were negative for MPO (d). The blasts also expressed CD2, CD5 (e), and were double positive for CD4 and CD8 (f). They were also positive for CD1a (g) and TdT (h), and negative for B-cell (CD19) and myeloid (MPO) antigens
Table 5.5
T-ALL Immunophenotype associated with T-cell maturation and differentiation
Stage | Immunophenotype |
|---|---|
Pre-thymic stage | Common lymphoid progenitor cell (pro-T) expressing CD34, TdT and HLA-DR and are negative for cytoplasmic CD3 (cCD3) |
Subcupsular cortical stage | Early T-cell precursors (pre-T) that express CD2, cCD3, CD7, TdT and are double negative (CD4- and CD8-). TCR gene at germ line configuration |
Cortical stage | Common thymocyte phenotype: CD1a, CD2, cCD3, CD7, TdT and double positive for CD4 and CD8. TCR gene rearrangement identified |
Medullary stage | Mature medullary thymocytes loose expression of TdT and CD1a, and express both cytoplasmic and surface CD3 and either CD4 or CD8 (single positive thymocytes) |
B Lymphoblastic Leukemia/Lymphoma
Clinical Features and Epidemiology
B lymphoblastic leukemia/lymphoma, in contrast to its T counterpart, usually has limited tissue involvement at presentation [56, 62]. The majority of patients present with peripheral blood and bone marrow disease, with skin, testes, and lymph nodes being among the most common sites of extramedullary involvement. Various molecular/cytogenetic B lymphoblastic leukemia/lymphoma groups are recognized in the WHO classification (Table 5.6).
Table 5.6
B-ALL with recurrent genetic abnormalities (2008 WHO classification)
Translocation | Fusion product | Frequency (%) | Prognosis | Immunophenotype |
|---|---|---|---|---|
t(9;22)(q34;q11.2) | BCR-ABL1 | 2–4 % in children | Poor | CD25+, CD117−, CD13+, CD33+, CD10+, CD19+, TdT+, CD34+, CD9+ |
t(v;11q23) t(4;11)(q21;q23) t(19;11)(p13;q23) t(9;11)(p22;q23) | MLL AF4-MLL ENL-MLL AF9-MLL | Most common Also seen in T-ALL Common in AML | Poor | CD19+, CD10−,CD24−, CD15/65+, CD9+ |
t(12;21)(p13;q22) | TEL-AML1 (ETV6-RUNX1) | 25 % of cases, common in children | Favorable | CD34+, CD13+, CD9− |
Hyperdiploidy >50 chromosomes (most common +21, +X, +14, +4) | 25 % of cases, common in children | Favorable | CD34+, CD45− | |
Hypodiploidy <46 chromosomes | – | <45 chromosomes 1 % | Poor | |
t(5;14)(q31;q32) | IL3-IGH@ | <1 % | Unknown | Eosinophilia |
t(1;19)(q23;p13.3) | E2A-PBX1 (TCF3-PBX1) | 6 % | Improved with current intensive therapies | CD9 + strong, CD34−, cμ heavy chain |
Imaging Features
Extensive thoracic or abdominal involvement is uncommon with B lymphoblastic leukemia/lymphoma. Accordingly, the use of FDG PET imaging for staging is generally not indicated. Typically, the initial staging evaluation is directed by symptoms and physical exam findings. For example, the presence of a testicular mass noted by ultrasound might prompt additional evaluation (Fig. 5.10).
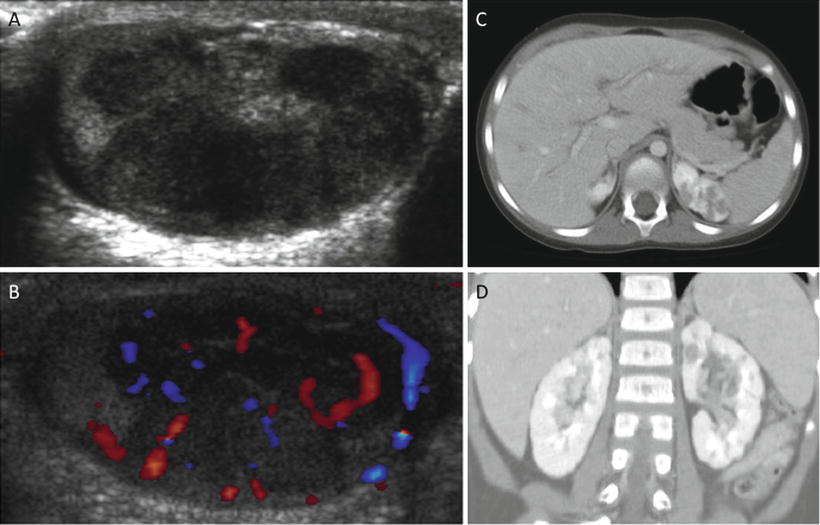
Fig. 5.10
Pre-B lymphoblastic lymphoma: 4-year-old with bilateral asymmetric, nontender testicular enlargement detected by pediatrician at a routine check-up. (a, b) US showed multifocal testicular masses with increased color Doppler flow (b). (c, d) CT showed additional bilateral renal involvement, L > R. Biopsy revealed pre-B cell lineage lymphoblastic lymphoma; bone marrow had <5 % blasts
Pathology
The morphologic features of B lymphoblastic leukemia/lymphoma are identical to those of T lymphoblastic leukemia/lymphoma. The B-cell lineage is defined by the expression of CD19 and one or more of the following markers: CD79a, cytoplasmic CD22 and/or CD10 (Fig. 5.11). In addition, the neoplastic cells are often positive for CD34 and TdT. PAX5 is a highly specific B-cell marker that is expressed in virtually all cases of B lymphoblastic leukemia/lymphoma [63]. It should be noted that the neoplastic cells of B lymphoblastic leukemia/lymphoma might be negative for CD20 and CD45 by immunohistochemistry. B lymphoblastic leukemia/lymphoma can be further subclassified into groups representing stages of B-cell maturation and differentiation (Table 5.7).
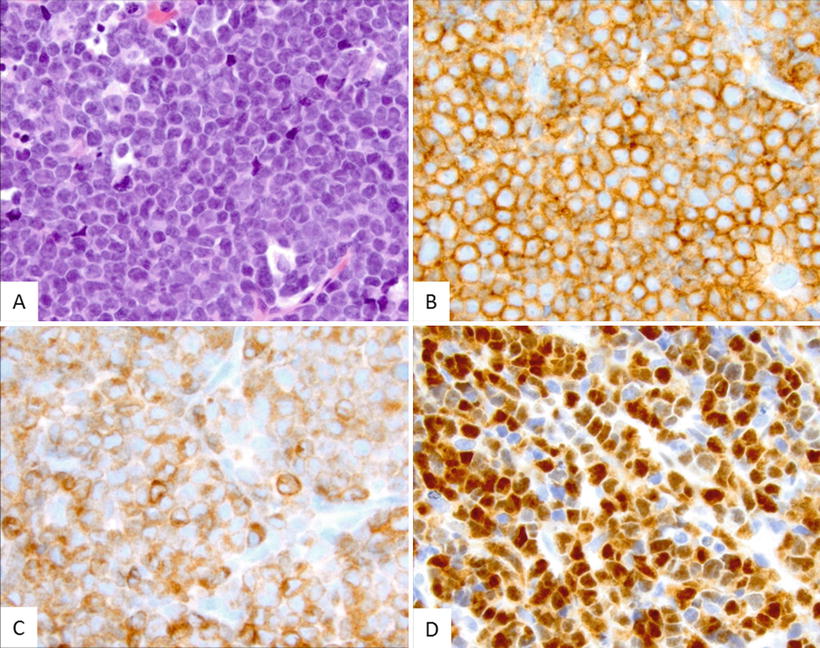
Fig. 5.11




This case of precursor B-cell ALL reveals a neoplastic infiltrate that is morphologically identical to precursor T-cell ALL. (a) Immunohistochemical stains showed that the blasts expressed characteristic B-cell markers being positive for CD19 (b) and PAX5 (c), as well as for TdT (d)
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
