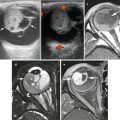
Fig. 12.1
Three-dimensional CT reconstruction of an abdominal neuroblastoma in a 4-year-old boy. Segmentation of the neuroblastoma (in green) from the anterior pancreas (yellow), the posterior kidneys (orange–brown), and aorta and its encased branches is displayed in three dimensions. This also allows computer calculation of the tumor volume (167 cc)
Neuroblastoma and Peripheral Neuroblastic Tumors
Definition: Neuroblastoma is an embryonal cancer of the sympathetic nervous system and primarily arises early in childhood. Neuroblastoma is one of the few human malignancies known to demonstrate spontaneous regression from a poorly differentiated malignancy to a completely benign one [11]. The etiology of neuroblastoma is unknown. A subset of patients that inherit a genetic predisposition to neuroblastoma may have multifocal tumors that arise at a very early age or are detected during screening ultrasound examinations.
Clinical Features and Epidemiology
Neuroblastoma accounts for 7–10 % of all childhood cancers, and it is the most common extracranial cancer diagnosed during infancy [12]. The annual incidence of neuroblastoma is 10.5 per million in children less than 15 years of age [13, 14]. The prevalence is about one case in 7,000 live births, and there are about 700 annual new cases in the United States [15]. Mass screening for neuroblastoma at 6–12 months of age led to an increased prevalence of neuroblastoma in the screened populations but no decrease in mortality for this disease, as the tumors detected had overwhelming favorable genetic features [15]. There is no strong evidence for epidemiological differences according to ethnical origin, although African Americans are more likely to have high-risk disease with fatal outcome [16].
Neuroblastoma occurs slightly more frequently in boys than girls, and the median age at diagnosis is 17 months [14]. There is no significant geographical variation in the incidence between North America and Europe, and there are no differences between races [16]. Some patients inherit a genetic predisposition to neuroblastoma due to germline mutations, whereas most appear to be caused by sporadic mutations [17]. Cases of familial neuroblastoma may have bilateral adrenal or multifocal primary tumors [15].
Clinical presentation of neuroblastoma depends upon site of origin, disease extent, and the presence of paraneoplastic syndromes (Table 12.1). The majority (65 %) arise in the abdomen, with most arising in the adrenal gland. Additional sites of origin include the neck, chest, and pelvis. There is a concordance with age and site of disease, with infants more likely to present with thoracic and cervical primaries [16]. Approximately one-half of patients present with localized or regional disease, and approximately 35 % have regional lymph node spread at the time of diagnosis [16]. Patients with localized disease are often asymptomatic, with disease coincidently diagnosed after testing for unassociated medical conditions. Alternatively, a mass or abdominal distension and pain are present. An abdominal neuroblastoma may present as diarrhea owing to secretion of vasoactive intestinal peptides. Patients who have localized cervical disease arising from the superior cervical ganglion may present with Horner syndrome. Epidural or intradural extension of tumors occurs in approximately 5–15 % of neuroblastoma and may be accompanied by neurologic impairment [16].
Eponym | Syndrome features |
|---|---|
Pepper syndrome | Massive involvement of the liver by metastatic neuroblastoma |
Horner syndrome | Unilateral ptosis, myosis, and anhydrosis associated with a thoracic or cervical primary tumor |
Hutchinson syndrome | Limping and irritability in young child associated with metastasis of neuroblastoma to bone and bone marrow |
Opsoclonus myoclonus ataxia syndrome | Myoclonic jerking and random eye movement with or without cerebellar ataxia, likely immune mediated. It may not resolve with tumor removal, and often exhibits progressive neuropsychological sequelae |
Kerner–Morrison syndrome | Intractable secretory diarrhea due to tumor secretion of vasointestinal peptides |
Neurocristopathy syndrome | Neuroblastoma associated with other neural crest disorders, including hypoventilation syndrome or Hirschsprung disease |
Blueberry muffin syndrome | Multiple blue/purple skin marks or nodules in the skin |
Raccoon eyes | Periorbital swelling and ecchymoses in neuroblastoma spread to periorbital region |
Most neuroblastomas secrete catecholamines, whose urinary metabolites include dopamine, vanillylmandelic acid, and homovanillic acid. These analytes are useful both for diagnosis and follow-up. Because infants with neuroblastoma have a better outcome than children who are more than 1 year at diagnosis, mass screening for neuroblastoma was pioneered by Japanese investigators who demonstrated that asymptomatic tumors could be detected in infants by measurement of urine catecholamines. Subsequent studies have shown that mass screening for neuroblastoma rarely detects biologically unfavorable tumors early and has not improved the overall mortality rate [15].
Imaging Features
Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma are sympathetic nervous system tumors of varying maturity derived from the primordial neural crest cells and may be found in the adrenal medulla (35–40 %) and extra-adrenal sites. In neuroblastoma and ganglioneuroblastoma, extra-adrenal sites include the retroperitoneum (30–35 %), posterior mediastinum (20 %), neck (1–5 %), and pelvis (2–3 %), in descending order of frequency [18]. Rarely, primary neuroblastoma can be found in the central nervous system (supratentorial primitive neuroectodermal tumor, see CNS Chap. 6 for detailed discussion) and within the kidney (mimicking mesoblastic nephroma [19]). In contrast, posterior mediastinum is the commonest site (41.5 %) for ganglioneuroma, followed by retroperitoneum (37.5 %), adrenal gland (21 %), and neck (8 %). The differentiation among the various histologic types by imaging is not accurate unless one finds metastases, which are only rarely reported in ganglioneuroma. In general, ganglioneuroma is found in older patients and is not as common as neuroblastoma. While the majority of neuroblastomas and ganglioneuroblastomas have irregular contours and sometimes show invasion of adjacent structures, most ganglioneuromas are round, well-demarcated, and occasionally lobulated in outline. Distant metastases from neuroblastic tumors, which may be present in 60–70 % of children at the time of presentation, are located in cortical bone (55 %), bone marrow (70 %), regional lymph nodes (30 %), liver (Pepper syndrome), skin, and rarely lung and brain [20, 21]. Patients with marrow infiltration have a better prognosis than those with cortical metastases.
Since patients can present with a large range of symptoms and signs and tumors occurring in various locations [22], the initial imaging modality may range from plain radiography to ultrasound, or other cross-sectional modalities with or without the use of ionizing radiation. It is important to be familiar with imaging findings of each modality used initially and the value of additional imaging modality to obtain further anatomical and functional information. The choice of modality also depends on physician preference and availability of local expertise and of imaging equipment or radionuclide.
Plain Radiography: Plain radiography of neuroblastic tumors may show evidence of a primary mass in the neck, posterior mediastinum (Fig. 12.2), retroperitoneum, or pelvis. Calcification can be detected in at least 30 % of neuroblastomas (Fig. 12.3). Lateral radiographs of neck tumors may show a prevertebral soft tissue mass with tracheal or pharyngeal displacement. In patients presenting with Horner’s syndrome, a mass may be seen in the lower neck or superior mediastinum. Posterior mediastinal tumors may cause rib erosion or splaying, lung compression, and displacement of the paraspinal lines from the adjacent mass effect (Fig. 12.4). The pedicles may be eroded and intervertebral foramina widened from intraspinal extension. Abdominal and pelvic masses may cause displacement of air-filled bowel loops. Paravertebral widening and increased interpediculate distances from intraspinal extension may be seen. Metastases to the skull (calvaria and skull base) and periorbital region appear as lucent (and less often sclerotic) destruction and ill-defined sutural diastasis from dural involvement [21]. Long bone metastases manifest as metaphyseal lucent erosions and periosteal new bone formation. Enlarged liver shadow may be seen in metastatic neuroblastoma to liver.
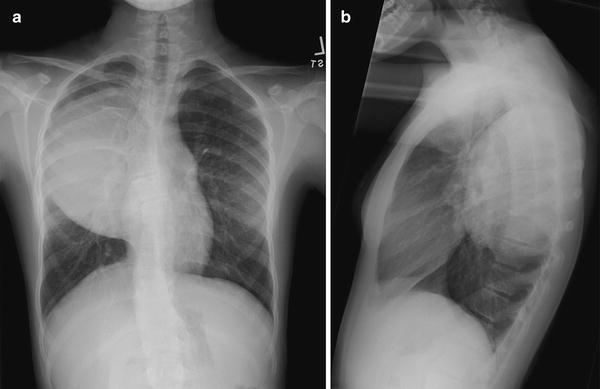
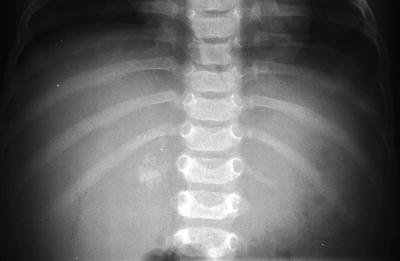
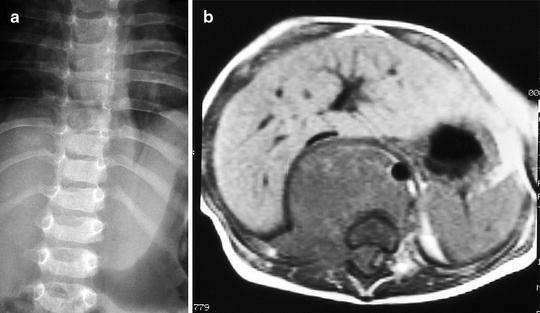
Fig. 12.2
Posterior mediastinal ganglioneuroma in a 12-year-old girl. Frontal (a) and lateral (b) views of the chest show a large soft tissue mass in the right upper posterior mediastinum with splaying of ribs. Histology of the excised mass showed a mature ganglioneuroma
Fig. 12.3
Right adrenal calcification in a 6-month-old girl with stage IVs adrenal neuroblastoma. Plain radiograph shows amorphous calcification in the region of right adrenal gland at the last thoracic spine level
Fig. 12.4
Paravertebral soft tissue mass with displacement of the left paravertebral stripe (a) in a 14-month-old boy with neuroblastoma. Enhanced axial (b) MRI of the thoracolumbar spine show retrocrural mass and, more clearly, right intraspinal extension
Ultrasound: Sonography is able to detect primary (usually adrenal and cervical) neuroblastic tumors and metastases (such as liver) in fetuses during obstetric ultrasound evaluation, mostly after 32 gestational weeks [23, 24]. Approximately 90 % of prenatally detected neuroblastomas are adrenal in origin, and the remainder either thoracic or cervical. Two-thirds of the adrenal tumors are on the right side. While solid masses tend to increase in size with serial antenatal ultrasound examinations, the cystic or complex ones may either grow or shrink on follow-up [25]. Ultrasonography (including use of color-coded and power Doppler technique) is useful in the differentiation between neonatal adrenal hemorrhage and neuroblastic tumors affecting the adrenal glands [26, 27]. A short-term follow-up ultrasound examination in 4–6 weeks showing resolution or decrease in size of mass and cystic or calcific changes is the most important differentiating factor favoring hemorrhage (Fig. 12.5). In neuroblastomas, a network of microscopic vessels supplying the tumor shows high velocity Doppler shifts; in adrenal hemorrhages, there is either decreased or absent flow to the mass. Neuroblastic tumors that undergo spontaneous regression and histologic maturation usually occur in stage 4S tumors and resolve in 6–12 months.
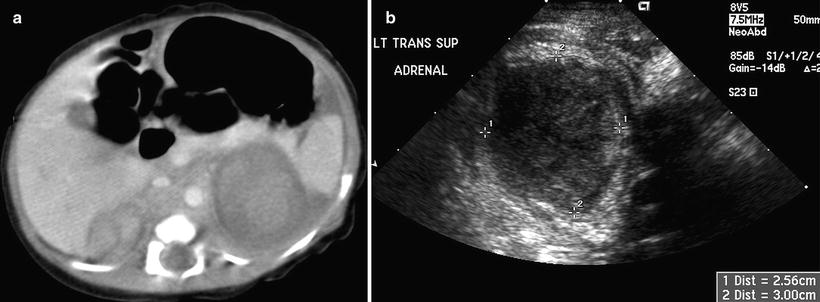
Fig. 12.5
Bilateral adrenal hemorrhage in a neonate. Contrast-enhanced axial CT scan (a) of the upper abdomen shows bilateral suprarenal masses with soft tissue densities. Transverse ultrasound image (b) shows an inhomogeneous hypoechoic mass in the suprarenal region. Both masses decrease in size on a 6-week ultrasound follow-up examinations
A subdiaphragmatic extralobar pulmonary sequestration is an important differential diagnosis (Fig. 12.6). It is purely hyperechoic or hypoechoic, mostly on the left side, and is less frequently seen than neuroblastoma. Finding an arterial supply from the thoracic aorta and stability of size are helpful in the differentiation from neuroblastoma. Other differential diagnoses of a suprarenal mass on prenatal ultrasound include a duplex collecting system with dysplastic changes, exophytic Wilms’ tumor, mesoblastic nephroma [19], retroperitoneal teratoma, liver tumor, and splenic cyst [24].
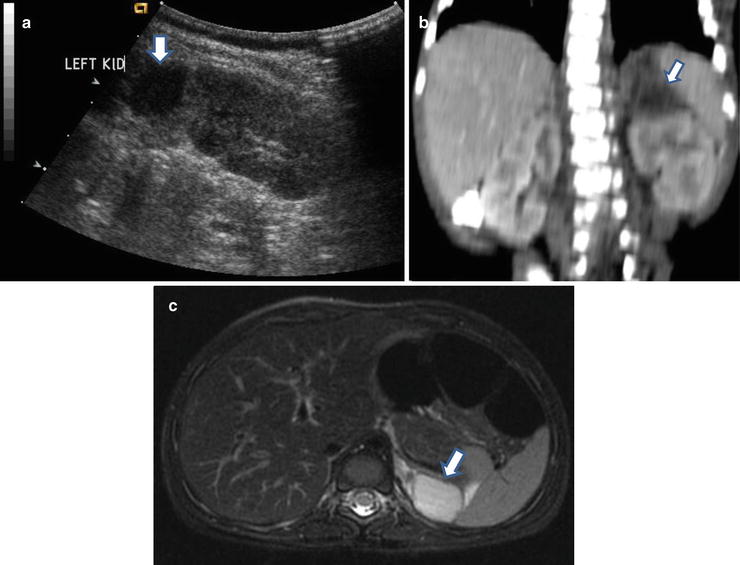
Fig. 12.6
Subdiaphragmatic extralobar pulmonary sequestration in a neonate. Antenatal ultrasound (not shown) shows a cystic mass in the retroperitoneum. Postnatal longitudinal ultrasound (a) of the abdomen shows a hypoechoic mass (arrow) just above the left kidney. Coronally reconstructed CT (b) of the abdomen shows a hypodense mass (arrow) indenting on the upper pole of left kidney. Axial T2W MR of the abdomen (c) shows a hyperintense mass (arrow) in the left suprarenal region. Histology of the excised specimen shows extralobar pulmonary sequestration
Neuroblastic tumors in the pediatric abdomen and pelvis appear as solid masses of varying echogenicity and heterogeneity (Fig. 12.7). Hyperechoic areas correspond to calcifications and cystic areas correspond to hemorrhage or necrosis. Calcifications may produce acoustic shadowing that limits assessment of the size and full extent of the tumor. Tumor of adrenal origin typically produces inferolateral displacement of the adjacent kidney. Hydronephrosis may be seen from obstruction of the collecting system by tumor or metastatic nodes. Real-time demonstration of differential movement between the mass and adjacent organs, such as liver and kidney, helps in the determination of the organ of origin or invasion of adjacent organ. Although ultrasonography may detect metastasis to liver and lymph nodes, or spread to pleural space, and rarely, scrotum [28, 29], it is not possible to detect extradural intraspinal extension of tumor. Duplex and color Doppler examination helps in evaluating blood vessels (aorta and inferior vena cava and their branches) that may be compressed, encased, or displaced by the tumor, or may demonstrate intravascular extension [27]. Ultrasound is also useful in follow-up, especially if an initial baseline examination is obtained.
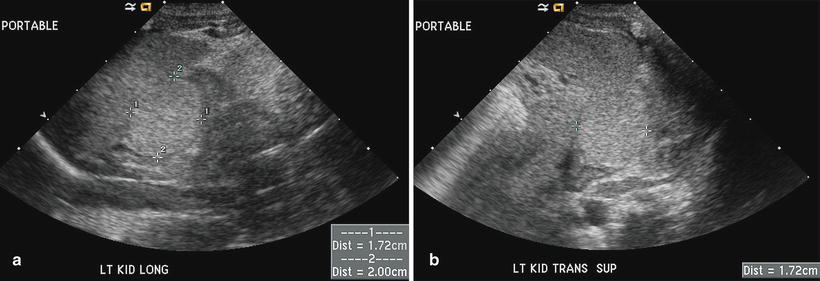
Fig. 12.7
Neuroblastic tumor in a newborn. A suprarenal mass was found incidentally on renal ultrasound performed for oliguria. Longitudinal (a) and transverse (b) ultrasound images show a uniformly echogenic mass measuring 1.7 × 1.7 × 2.0 cm in the left suprarenal fossa. The mass is MIBG avid, and it regresses spontaneously on follow-up ultrasound examination
Computed Tomography: Intravenous-enhanced CT is the most commonly used imaging modality in the assessment of origin and extent of neuroblastic tumors. Regions of coverage include chest, abdomen, pelvis, neck, and skull, depending on the location of the mass at presentation. Findings suggestive of a neuroblastic tumor include calcifications hemorrhage and/or necrosis, lymphadenopathy, vascular involvement (Fig. 12.8), and metastases to liver (Fig. 12.9) and bone. Metastases to lung are infrequent (1–3 %). Most masses are heterogeneous, with fine or coarse calcifications (80–90 % of neuroblastoma and ganglioneuroblastomas and 40–60 % of ganglioneuromas). Calcifications are usually coarse, mottled, and amorphous in neuroblastomas and discrete and punctate in ganglioneuromas. Ganglioneuroma may show a delayed and progressive heterogeneous enhancement pattern [30]. Hemorrhage and/or necrosis are more common in larger tumors, appearing as area of low attenuation. Lymphadenopathy is commonly seen around the renal hilum (Fig. 12.10), liver hilum, retroperitoneum. It is less common in the neck (Fig. 12.11) and mediastinum. When the primary is large, it is often difficult to differentiate between the primary and metastatic nodes. Vascular displacement, encasement, or invasion may be seen (Fig. 12.10). In the abdomen, the aorta and the inferior vena cava and their branches are commonly involved. Right atrial extension of tumor thrombus has been reported [31]. Tumor arising from the adrenal gland produces displacement of the adjacent kidney laterally and caudally, best demonstrated with a coronally reconstructed image. Up to 20 % of neuroblastomas have been shown to invade the renal parenchyma and these tumors tend to be extensive, of unfavorable histological features, and involve lymph nodes. CT scan was found to be sensitive and specific for showing renal parenchymal invasion [32]. Extra-adrenal retroperitoneal or posterior mediastinal masses (Fig. 12.12) may also invade the paraspinal muscles or extend into the intraspinal epidural space via the neural foramina. Paraspinal tumors may displace the kidney anteriorly. Retrocrural extension is well demonstrated by CT. Cervical neuroblastic tumors are uncommon, usually presenting as a mass in the parapharyngeal space, displacing or involving the adjacent carotid artery and jugular vein (Fig. 12.11). Pelvic neuroblastic tumors arise from the presacral space and the anteriorly located organ of Zuckerkandl (Fig. 12.13). CT allows demonstration of bony metastases as lytic destruction or erosions. In the skull, additional dural involvement is associated with enhancing dural masses indenting underlying brain; extension into a suture is associated with sutural diastasis (Fig. 12.14). “Raccoon-eye” discoloration and proptosis may be the first presenting signs of involvement of the sphenoid bone and periorbital regions (Fig. 12.15). Rare brain metastasis can be intraventricular, leptomeningeal, or parenchymal in location [21].
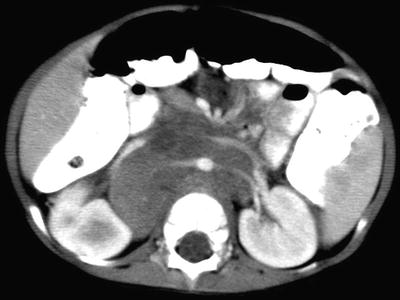
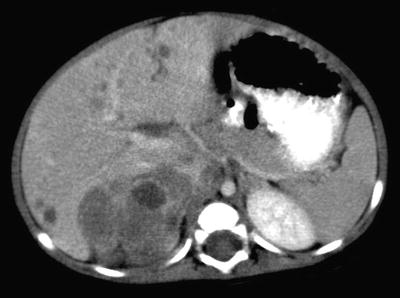
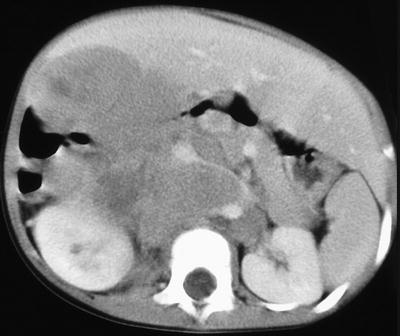
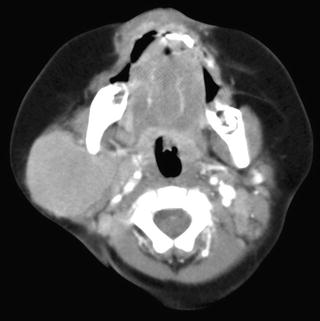
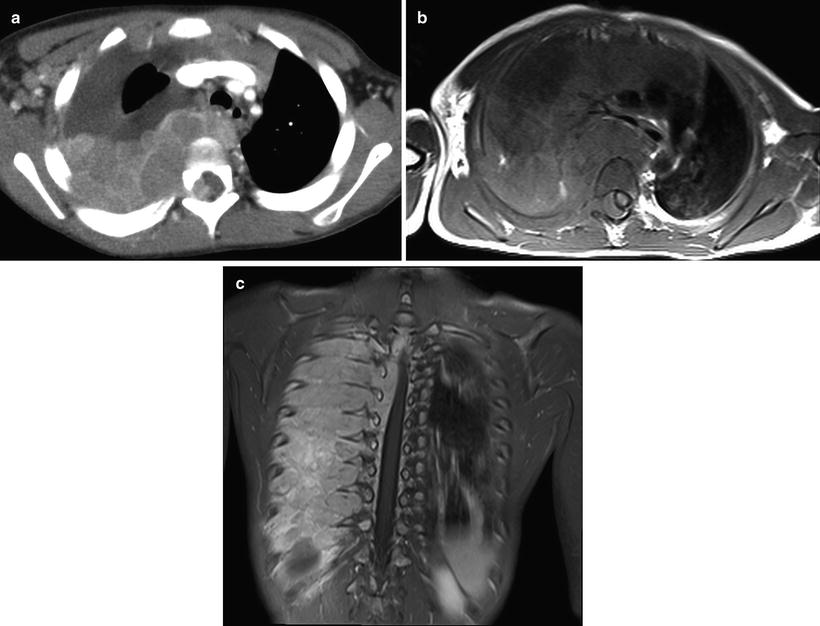
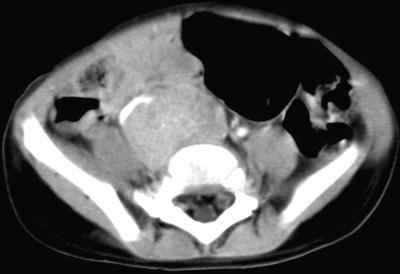
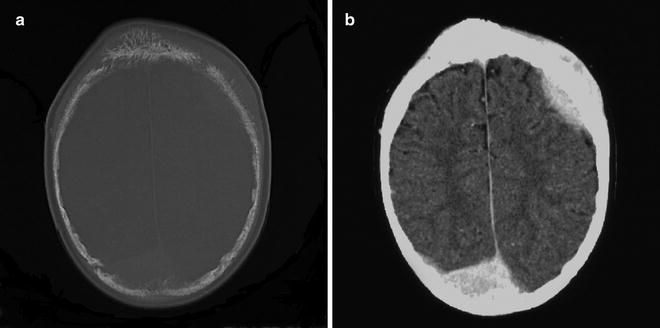
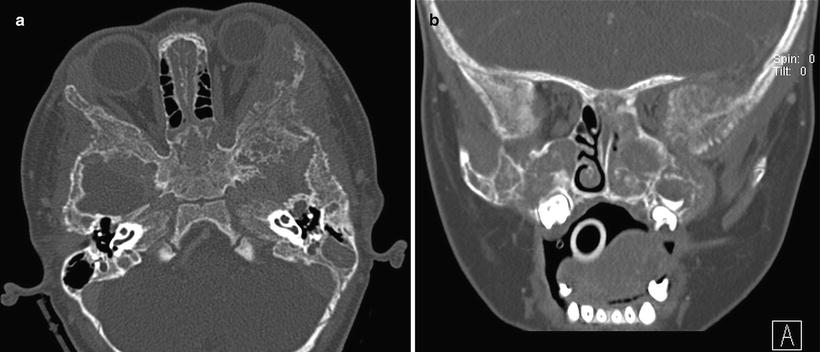
Fig. 12.8
Right adrenal neuroblastoma in a 2-year-old girl with vascular encasement and displacement. Contrast-enhanced CT scan of the upper abdomen shows a right adrenal tumor with retroperitoneal lymphadenopathy encasing abdominal aorta and renal vasculature, anterior displacement of the inferior vena cava without intravascular invasion, and extension over the vertebral body without invasion
Fig. 12.9
Metastatic neuroblastoma to liver from a right adrenal neuroblastoma in a 7-month-old boy. Contrast-enhanced axial CT image of the liver shows multiple small hypodense lesions in both lobes of the liver. A right adrenal mass with hypodense necrotic or hemorrhagic areas is also seen
Fig. 12.10
Large neuroblastoma in the retroperitoneum in a 30-month-old girl. Axial CT image of the upper abdomen shows the relation of the mass to the hilar lymph nodes, right lobe of liver, both kidneys, aorta, inferior vena cava, renal vasculature, and vertebral body
Fig. 12.11
Metastatic cervical neuroblastoma from a right adrenal neuroblastoma in a 7-month-old boy. Axial CT image of the head and neck shows a soft tissue mass in the right parotid space. Biopsy of the neck mass showed neuroblastoma. An abdominal CT showed a right adrenal neuroblastoma with liver metastases (not shown)
Fig. 12.12
Thoracic neuroblastoma with intraspinal extension in a 34-month-old boy. Intravenous enhanced axial CT (a) image shows an enhancing right posterior mediastinal mass crossing midline with intraspinal extension. Enhanced axial (b) MR image shows the similar finding with displacement of the spinal cord to the left. The coronal (c) MR image shows a large posterior mediastinal mass with extensive chest wall and intraspinal involvement through intervertebral foramina at multiple levels
Fig. 12.13
Differentiating neuroblastoma of the organ of Zuckerkandl in a 17-month-old boy. Enhanced axial CT image of the abdomen shows a mass in the lower abdomen and upper pelvis with anterior displacement of the right iliac artery. Fine calcifications are present in the mass.
Fig. 12.14
Metastatic poorly differentiated neuroblastoma to skull with associated dural masses in a 9-year-old boy. Enhanced CT image of the skull with bone window (a) shows erosive skull changes close to sutures and associated enhancing dural deposits when displayed on brain window (b)
Fig. 12.15
Metastatic neuroblastoma to bilateral periorbital region in a 14-month-old girl presenting with bilateral proptosis and bruising. Axial (a) and coronal (b) CT images of the orbits show bilateral erosions of the lateral orbital walls as well as the facial and sphenoid bones
Magnetic Resonance Imaging: Magnetic resonance imaging is an important imaging modality of choice because it does not utilize radiation and provides high contrast resolution for the detection, multiplanar localization, and assessment of the extent of primary tumor involvement [33]. It is superior to CT to assess marrow infiltration and intraspinal extension. MRI is useful to evaluate resectability, to detect recurrence and metastasis, and to assess response to treatment and development of complications [34]. While the coronal plane is optimal to define the origin of organ involvement, spinal canal extension, and abdominal great vessel involvement, the axial plane (Fig. 12.16a, b) is the best to assess involvement of the origins of celiac axis and superior mesenteric artery [34]. Compared to CT, T1W images provide superior anatomic resolution and T2W images provide superior contrast differentiation from normal tissues. The multiplanar capability (especially coronal and sagittal planes, Fig. 12.16c, d) improves determination of vascular invasion and, therefore, resectability. The definition of the location of tumor mass in relation to the hemidiaphragms or crura is useful to determine the surgical approach (infra- or supradiaphragmatic). In the abdomen, the aorta and inferior vena cava are usually displaced anteriorly, and the various branches from these vessels may be encased (Fig. 12.16) or invaded (Fig. 12.17). Retroperitoneal lymphadenopathy and spread to various intra-abdominal organs may be seen. Differentiation from renal tumors, adrenal hemorrhage, and lymphoma is facilitated with the multiplanar depiction. MR imaging was found to be the single best test for detection of nodal and chest wall involvement (Figs. 12.12b, c) and intraspinal extension for thoracic neuroblastoma [35]. MR imaging shows more accuracy than CT for the detection of stage 4 disease, with sensitivities of 83 and 43 % (p < 0.01) and specificities of 97 and 88 % (p > 0.05), respectively [36], and it could replace the combination of CT and bone scintigraphy for overall assessment of stage 4 disease in children with neuroblastomas [36].
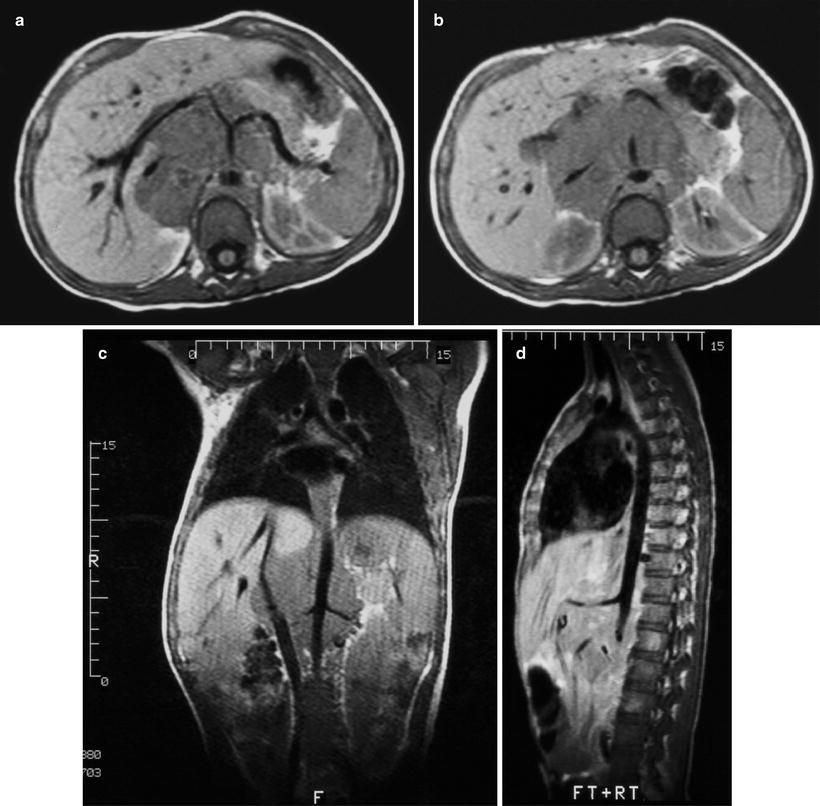
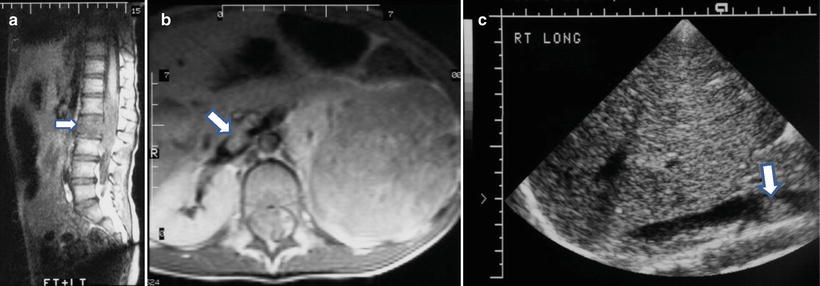
Fig. 12.16
Retroperitoneal neuroblastoma in a 13-year-old boy. Axial T1W MR images of the upper abdomen at the levels of the celiac axis (a) and the superior mesenteric artery (b) show tumor encasement of the abdominal aorta, celiac axis and its branches, the superior mesenteric artery, and the inferior vena cava. There is anterior displacement of the main portal vein. Coronal (c) and sagittal (d) images provide improved determination of vascular relationship to tumor. Encased renal vessels are noted
Fig. 12.17
Left neuroblastoma in a 5-year-old girl with lumbar vertebral marrow involvement and extension into inferior vena cava and spinal canal. Sagittal T1W image (a) shows tumor replacement of fatty marrow (arrow) and intraspinal extension around the conus medullaris. Axial post-contrast T1W MR (b) and longitudinal ultrasound (c) images show a large left suprarenal mass with a tumor thrombus (arrow) in the inferior vena cava. Intraspinal extension is also seen in (a) and (b)
Without intravenous contrast, neuroblastomas and ganglioneuroblastomas are typically heterogeneous with low signal intensity on T1W images and high signal intensity on T2W images. Ganglioneuromas are typically homogeneous but with T1W and T2W signal intensities similar to other neuroblastic tumors (Fig. 12.18). Hemorrhagic areas may show increased signal intensity on T1W images; cystic areas will appear bright on T2W images. Although calcifications may appear dark, detection may be difficult as compared to CT or sonography. After administration of intravenous contrast, neuroblastoma and ganglioneuroblastoma show heterogeneous enhancement while ganglioneuromas show variable and delayed enhancement [37]. In tumors contiguous with the kidneys, the delineation of the mass from the kidney may be lost when the enhancement of the tumor and the adjacent kidney parenchyma is to the same degree. MRI provides the best multiplanar assessment of intraspinal extension (dumbbell tumors) of paraspinal neuroblastic tumors, irrespective of the level of spinal involvement (10 % abdominal and 28 % intrathoracic). The post-contrast images will depict the enhancing intraspinal epidural tumor and assess its relation to the displaced or compressed spinal cord or nerve roots at neural foramina. Dural involvement associated with cranial vault metastasis shows enhancement of the soft tissues around the bony destruction (Fig. 12.19). Involvement of bone marrow by neuroblastic tumor will appear as low signal intensity on T1W images and high signal intensity on T2W images (Fig. 12.17). Two patterns of bone marrow involvement have been described. A nodular pattern is associated with less cortical metastases and a better prognosis. A diffuse pattern is associated with cortical metastases and is less responsive to chemotherapy [38]. MRI, however, is not useful to assess marrow tumor response to treatment since signal abnormalities may persist for a long time despite good response. Liver metastases may either be nodular or diffuse in distribution. Nodular metastases to liver often result in hepatomegaly and appear dark on T1W images and bright on T2W images. MRI is superior to CT in the depiction of diffuse liver metastatic infiltration in stage IVS disease (patients under 1 year of age with metastases to liver, skin, and bone marrow) as increased signal intensity on T2W images.
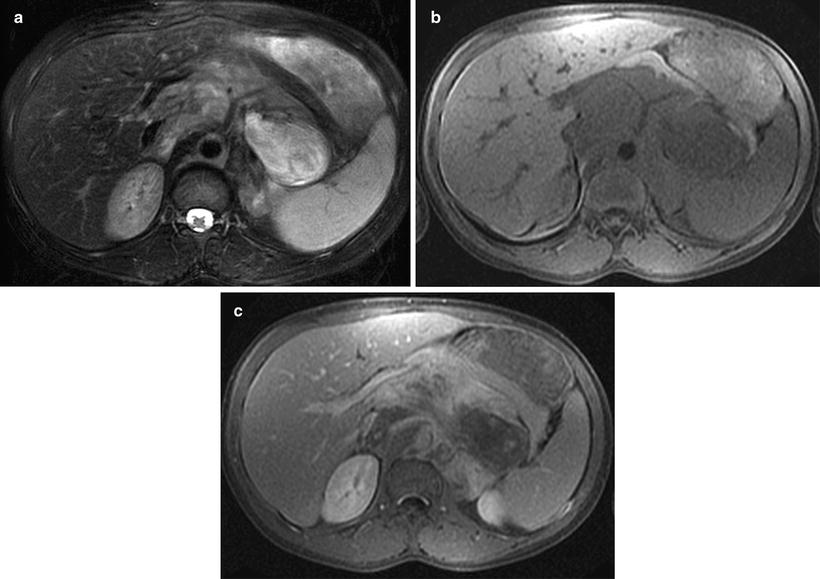
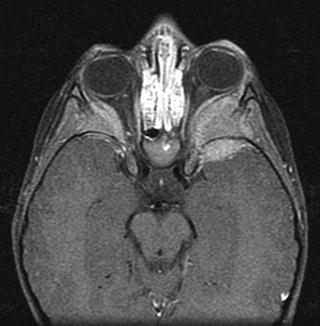
Fig. 12.18
Ganglioneuroma in a 13-year-old boy in the retroperitoneum. Fat-saturated T2W (a) and T1W (b) images of the upper abdomen showed retroperitoneal mass encasing abdominal vasculature with inhomogeneous bright and isointense signal intensities, respectively. Post-gadolinium fat-saturated T1W image (c) of the same region showed inhomogeneous enhancement of the tumor. Signal intensities in all sequences are indistinguishable from those with neuroblastoma. Histology showed mature ganglioneuroma
Fig. 12.19
Metastases to both lateral orbits in 3-year-old boy with a large left adrenal neuroblastoma. Enhanced MRI of the orbits shows bilateral soft tissue masses involving the lateral orbital walls and left greater wing of sphenoid bone with extradural extension into both middle cranial fossae
Diffusion-weighted sequences show increased signal intensity (restricted diffusion) in tumors owing to the high nuclear-to-cytoplasm ratio limiting intracellular motion and the densely packed tumor cells inhibiting effective motion of the extracellular water protons. Preliminary data showed that diffusion-weighted echo-planar imaging may be able to distinguish neuroblastoma from ganglioneuroblastoma or ganglioneuroma using the apparent diffusion coefficient (ADC). Neuroblastomas were found to have a lower ADC, reflecting their small densely packed, immature cells containing little cytoplasm versus the former groups with abundant cytoplasm surrounded by more stroma [39]. The potential value of ADC in assessing therapeutic response remains to be seen. Whole body MRI (WBMRI) using a STIR sequence has shown high sensitivity but low specificity in the detection of bony metastasis and has a low positive predictive value in comparison to I-123-MIBG scintigraphy [6, 8, 40].
Nuclear Medicine: Various radionuclide scintigraphic studies have been used in the localization of primary tumor and surveillance of metastases in patients with neuroblastic tumors [41]. In children with bone pain, whole body 99mTc-bone scintigraphy is used to detect bony cortical metastases, manifested typically as areas of increased radionuclide uptake, corresponding to destructive lesions on plain radiography (Fig. 12.20) [42]. Occasionally, photopenic “cold” foci may appear. Bone scintigraphy increases the sensitivity detecting bony metastasis by radiographic skeletal survey from 35–70 % to 90 % [43]. Unlike 123I-metaiodobenzylguanidine (MIBG) scintigraphy, bone scans can differentiate between cortical bone and bone marrow involvement, an important distinction in the staging of patients <18 months of age [41]. Involvement of the bony cortex has been shown to carry prognostic significance. Detection of bone involvement at long bone ends may be difficult owing to the relatively high normal uptake of bone-seeking agents in the growth plates in children. Over 60 % of neuroblastomas have increased uptake in the primary tumor. Even areas of tumor calcifications show increased uptake, this uptake may not correlate with extent of calcification or degree of necrosis [44]. Since up to 10 % of MIBG scans may be negative in neuroblastoma patients, bone scintigraphy and MIBG scans are complementary to each other. After therapy, areas with increased uptake on 99mTc-bone scintigraphy may persist for an extended period of time and with variable intensity.
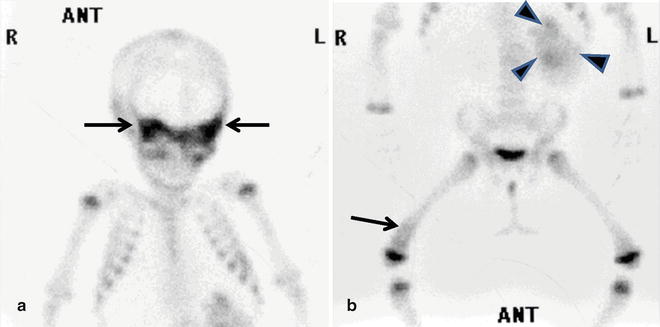
Fig. 12.20
Multiple bony metastases from retroperitoneal neuroblastoma in a 10-month-old girl. Anterior views of the body on skeletal scintigraphy (Tc-99m-MDP) shows increased uptake in (a) sphenoid bone (arrows), orbits, and (b) distal right femur (arrow). Note uptake of the radionuclide in part of the primary tumor (arrowheads)
Radiolabeled 123I-MIBG is a catecholamine analog taken up catecholamine-producing primary or metastatic tumors (bone, bone marrow, and soft tissues) and mainly excreted by the kidneys (Fig. 12.21). In addition to neuroblastic tumors, neuroendocrine tumors including pheochromocytoma and carcinoid tumors also show uptake of MIBG. MIBG is concentrated within the cytoplasm of neuroblastoma cells. Retention of MIBG is enhanced by its rapid reuptake of any MIBG that exits the tumor cells [41]. The guidelines for evaluation of the metastatic response and the extent of neuroblastoma using 123I-MIBG scans had been reported, using a semiquantitative scoring system (the Curie scoring system), based on the extent of metastases in each of the 9 anatomic sectors for osteomedullary and soft tissue lesions in the body [45, 46]. This agent has superior imaging quality and radiation dose properties (shorter half-life of 13.2 h, ideal photon energy of 159 KeV for gamma camera and single-photon emission computed tomography [SPECT], and lack of a beta particle) over 131I-MIBG, but there is no significant difference in survival predictability in patients with stage IV neuroblastoma by the type of scan [47]. Planar images are obtained most commonly at 24 h.
The addition of 48-h delayed images and tomographic technique (SPECT) has been shown to increase the number of detected lesions and to better anatomically localize neuroblastomas [48]. Fusion of SPECT data to separately acquired anatomical imaging studies (CT or MRI) can be performed [49] (Fig. 12.22). SPECT imaging is most useful in localizing lesions near areas of physiologic uptake, such as in the retroperitoneum. Normal sites of radiotracer distribution include the nasal mucosa, salivary glands, thyroid gland, the upper chest, myocardium, liver, normal adrenal tissues (especially after contralateral adrenalectomy), and the urinary and gastrointestinal tracts , the latter two resulting in false positives. False negatives (mostly bony metastases) may be caused by pharmacological interference from some commonly prescribed and readily available over-the-counter medicines [50], and possibly tumor cell differentiation and maturation [40]. About 57 % of ganglioneuromas accumulate MIBG, and these MIBG-avid tumors usually produce increased amount of urinary catecholamines.
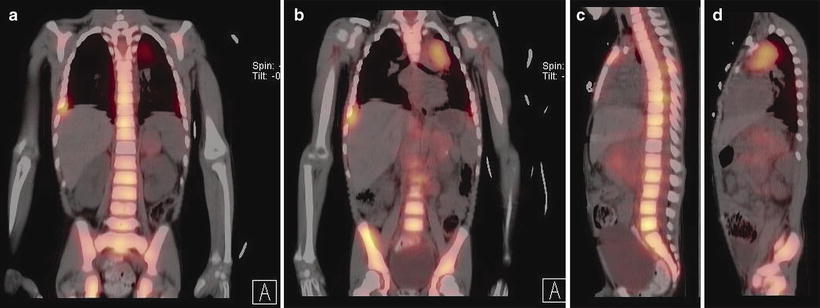
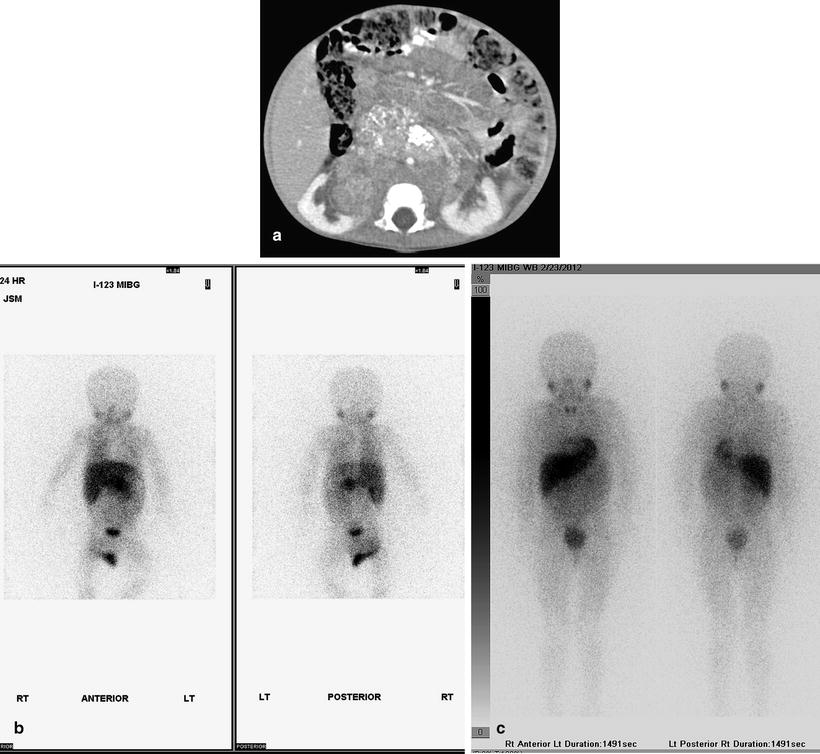
Fig. 12.21
Retroperitoneal neuroblastoma in an 11-month-old girl. Contrast-enhanced axial CT image (a) shows a large retroperitoneal neuroblastoma encasing abdominal vasculature and displacing kidneys. Increased uptake is present in part of the tumor shown on planar (b) I-123-MIBG scintigraphy. After chemotherapy and surgical treatment, follow-up axial CT (not shown) shows no residual tumor and planar I-123-MIBG scintigraphy (c) shows no abnormal uptake
Fig. 12.22
(a–d) Extensive metastatic neuroblastoma in a 4-year-old boy. Fusion of I-123MIBG SPECT and CT images reveals abnormal uptake in retroperitoneum, left upper mediastinum, and extensive metastases to the spine, sternum, scapula, and ribs
A recent meta-analysis of 123I-MIBG scintigraphy estimated a sensitivity of 97 % in the detection of neuroblastomas [51]. Detection of presacral neuroblastic tumors may be missed when there is a full urinary bladder distended with the excreted radionuclide. This pitfall can be reduced by catheterization of the urinary bladder during imaging. 123I-MIBG carries a relatively high radiation dose of 13–19 mSv. In patients with neuroblastoma presenting with Horner’s syndrome secondary to sympathetic denervation, there is asymmetric loss of normal uptake by the ipsilateral salivary glands. Although results of MIBG scan correlate fairly well with bone marrow aspirates and MRI, MIBG cannot replace marrow aspirates at diagnosis and evaluation of response in children with neuroblastoma. The combined use of 123I-MIBG scintigraphy and MRI has been shown to increase the sensitivity and specificity of the diagnosis of neuroblastoma [52]. Poor prognosis has been associated with MIBG-avid tumors in stage IV patients over 1 year of age at presentation and in those patients that remain avid after chemotherapy.
18F-FDG-PET scan has been shown to be equal or superior to MIBG scintigraphy for localizing neuroblastic tumors in soft tissue and extracranial skeleton, for identifying small lesions, and for delineating the extent of disease [53, 54]. 18F-FDG PET/CT may be useful in the <10 % of patients with primary neuroblastoma that are not 123I-MIBG avid or when there is suspicion that disease extent exceeds what is depicted with 123I-MIBG scan [55] (Fig. 12.23). Moreover, tumoral avidity (maximal standardized uptake value of the most intense soft tissue and bone-bone marrow lesion) for 18F-FDG and the extent of 18F-FDG-avid bone-bone marrow disease were identified as adverse prognostic factors in high-risk neuroblastoma patients undergoing high-dose 131I-MIBG therapy. In a study [56] comparing the diagnostic utility of 123I-MIBG (with SPECT) to 18F-FDG PET (or PET-CT) scans in neuroblastomas of all stages, the investigators found that 18F-FDG PET was superior in the follow-up of patients with stages 1 and 2 disease because the extent of tumor was better demonstrated relative to background and when bone marrow involvement was not likely to be present. Also, 18F-FDG PET scan better depicts disease sites in stage 3 and 4 patients when the tumor was weakly positive for 123I-MIBG. Moreover, 18F-FDG PET scan was useful in showing extent of disease in chest, abdomen, and pelvis when disease involvement on CT or MRI appeared more extensive than shown with 123I-MIBG scan. However, 123I-MIBG scan was superior in the evaluation of stage 4 neuroblastoma patients, primarily because of the better detection of bone or bone marrow metastases. Detection of these metastases with 18F-FDG PET scan was often difficult owing to the normal physiologic uptake pattern in the growing skeleton and skull metastases were masked by normal brain uptake. Also, bone and bone marrow lesions could be masked or falsely interpreted as positive in patients receiving granulocyte colony-stimulating factor. In another study comparing 123I-MIBG scintigraphy/SPECT and 18F-FDG PET scan in pediatric neuroblastomas [57], the sensitivities were 50 and 78 % and the specificities were 75 and 92 %, respectively. The combined use of both modalities increased the sensitivity to 85 %. False positive results of each modality were due to physiological uptake or post-therapy changes, and false negative results from low uptake and small lesion size. These authors suggest that 18F-FDG PET may be useful when there is a discrepancy between the 123I-MIBG and morphologic imaging or when there are inconclusive findings.
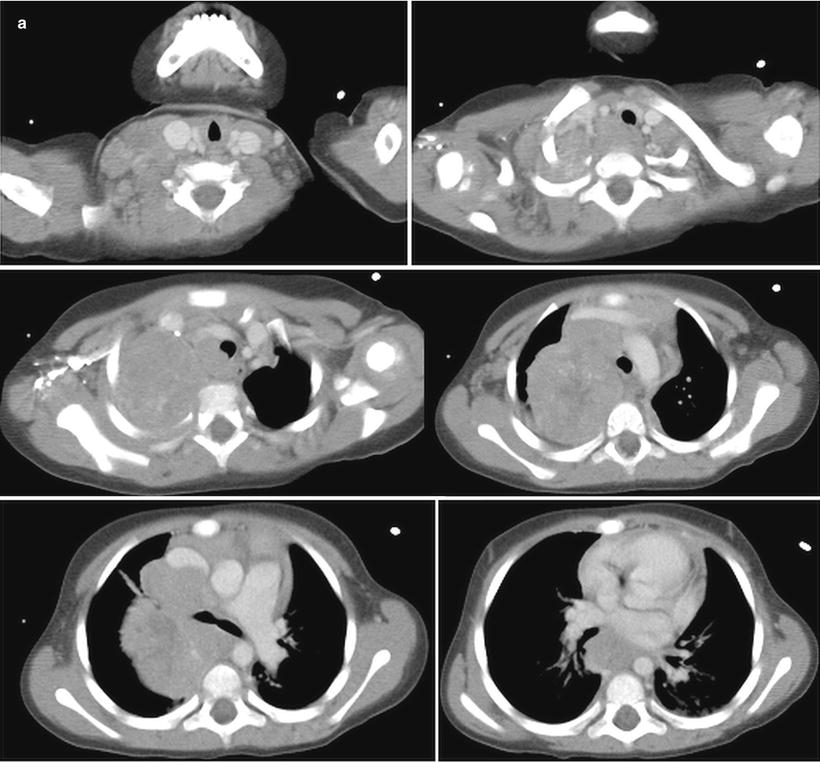
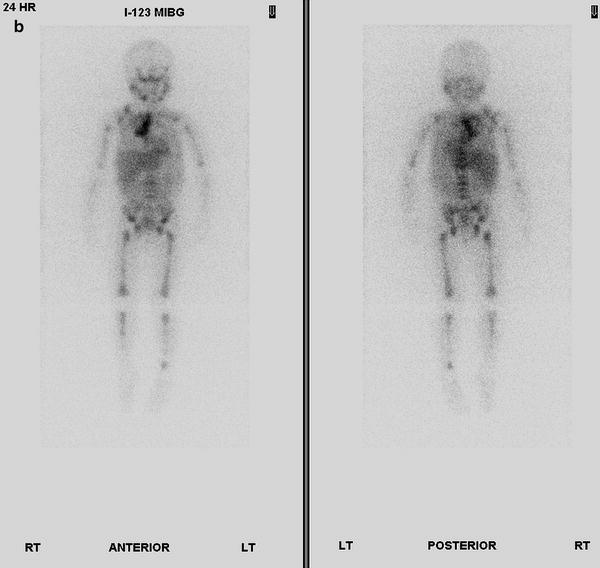

Fig. 12.23
PET-CT scan of thoracic neuroblastoma in a 35-month-old girl. Contrast-enhanced axial CT images (a) of the thorax show a large posterior and superior mediastinum mass. (b) I-123-MIBG scan shows abnormal uptake in right lung apex and mediastinum, right supraclavicular nodes, and bone marrow. Coronal 18F-FDG PET images (c) more clearly show areas of increased activity in the neck, both sides of mediastinum, and bone marrow of the axial and appendicular skeleton. Fusion of 18F-PET images to CT images is not shown
Molecular Genetics
A family history is observed in about 1 % of neuroblastoma cases [58]. Germline heterozygous mutations in the homeobox gene PHOX2B has been described in these patients [59]. The PHOX2B gene, localized at chromosome 4p12, regulates development of the autonomous nervous system [58]. Mutations in PHOX2B are associated with other neural crest disorders such as Hirschsprung disease and Ondine curse [59], and they also occur in rare sporadic neuroblastomas.
Activating mutations in the tyrosine kinase domain of the anaplastic lymphoma kinase (ALK) oncogene on chromosome 2p23 account for most cases of hereditary neuroblastoma [60]. Copy number increases of ALK have been found in 20–25 % of primary neuroblastomas, and elevated expression of ALK occurs in aggressive neuroblastoma.
Associations between neuroblastic tumors and various “neuro-cardio-faciocutaneous” syndromes, including Noonan and Costello syndromes, and neurofibromatosis type (NF1), are defined by the constitutive activation of the RAS-MAPK pathway [61] and have also been reported [62]. However, germline mutations of NF1, ALK, and PHOX2B do not account for all familial cases of neuroblastoma. Various types of abnormal constitutional karyotypes have been observed in neuroblastoma, including copy number anomalies, balanced and unbalanced translocations, and specific chromosomal deletions [60, 63]. NBPF1 (Neuroblastoma Break point Family) is another candidate neuroblastoma susceptibility gene [64], and NBPF23, another member of this gene family, is associated with neuroblastoma susceptibility [65]. Associations have also been found with single nucleotide polymorphisms at chromosome 6p22 within the putative FLJ22536 and FLJ44180 genes and at 2q35 within the BARD1 genes [66, 67].
Pathology
Currently neuroblastoma tumors are classified according to the International Neuroblastoma Pathology Classification (INPC) [68–70], which distinguishes between two prognostic groups, favorable histology (FH) and unfavorable histology (UH), according to the amount of Schwannian stroma, ganglionic differentiation, the mitotic and karyorrhectic index (MKI), and the age of the patients (Table 12.2). MKI should be assigned to each of the neuroblastoma tumor categories and comprises low (− <100/5,000 cells), intermediate (100–200/5,000cells), or high (>200/5,000 cells). Peripheral neuroblastic tumors are assigned to one of four basic morphological categories: neuroblastoma, nodular ganglioneuroblastoma, diffuse ganglioneuroblastoma, and ganglioneuroma [69, 71].
Age | Any age | <1.5 years | 1.5 to <5 years | ≥5 years |
|---|---|---|---|---|
Favorable histology | Ganglioneuroma (SSD) Maturing/mature GNB, intermixed (SSR) | Neuroblastoma (SSP) PD with low/intermediate MKI Differentiating with low/intermediate MKI | Neuroblastoma (SSP) Differentiating with low MKI | GNB Nodular (composite, Schwannian stroma-rich/stroma-dominant and stroma-poor), favorable subset |
Unfavorable histology | Neuroblastoma (SSP) Undifferentiated with any MKI | Neuroblastoma (SSP) PD with high MKI Differentiating with high MKI | Neuroblastoma (SSP) PD any MKI Differentiating with intermediate or high MKI | Neuroblastoma (SSP) with any subtype and any MKI GNB Nodular (composite, Schwannian stroma-rich/stroma-dominant and stroma-poor), unfavorable subset |
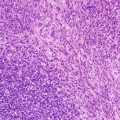
Neuroblastoma: Neuroblastoma (Schwannian stroma-poor) is composed of groups/nests of small dark primitive appearing tumor cells (neuroblasts) with dark nuclei, scant cytoplasm, and poorly differentiated cytoplasmic borders. The cells grow in solid sheets separated by thin fibrovascular septal tissue. S100 positive slender cells (putative Schwann cell blasts) can be detected in septa. Such tumors may be difficult to differentiate morphologically from other small round cell tumors. The neuroblastoma category consists of three subtypes, based on degree of neuroblastic differentiation: undifferentiated, poorly differentiated, and differentiating.
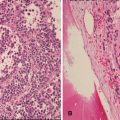
In undifferentiated neuroblastoma, virtually all the tumor cells are undifferentiated neuroblasts, and the tumor cannot be diagnosed without ancillary studies such as immunohistochemistry. Tumor cells may contain nucleoli, which has been found to be an unfavorable feature [72]. Neuropil is virtually absent. Neuronal markers are typically positive in only sporadic tumor cells. Poorly differentiated neuroblastoma, the most common subtype, shows ganglionic differentiation in less than 5 % of the tumor neuroblasts A variable amount of neuropil is easily recognized. Homer-Wright rosettes may be present (Fig. 12.24). In differentiating neuroblastoma, at least 5 % of the neuroblasts show definite ganglionic differentiation, and the lesion contains moderate to abundant neuropil.
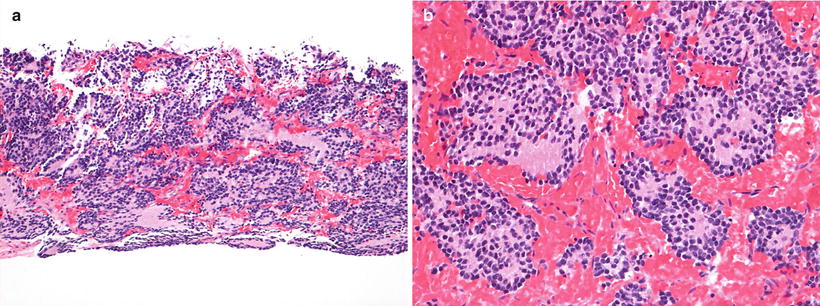
Fig. 12.24
Neuroblastoma poorly differentiated subtype (Schwannian stroma-poor). (a) Core biopsy shows neurites in the background and rosette formation (H-E10X). (b) Detail of Homer-Wright rosettes (H-E20X)
Ganglioneuroblastoma-intermixed (Schwannian stroma-rich) is a tumor with mixed maturation, typically a predominant ganglioneuromatous component (>50 %), and microscopic nests of neuroblasts and neuropil showing a variable degree of differentiation with or without the presence of ganglion cells. The ganglioneuromatous component contains a Schwann cell matrix containing mature ganglion cells.
Ganglioneuroblastoma-nodular (composite Schwannian stroma-rich/stroma-dominant and stroma-poor) is a composite and multiclonal tumor characterized by the presence of one or more grossly visible often hemorrhagic/necrotic nodules (neuroblastoma nodules) coexisting with ganglioneuroblastoma-intermixed or with ganglioneuroma nodules (Fig. 12.25). Grading should be based on the neuroblastomatous component.
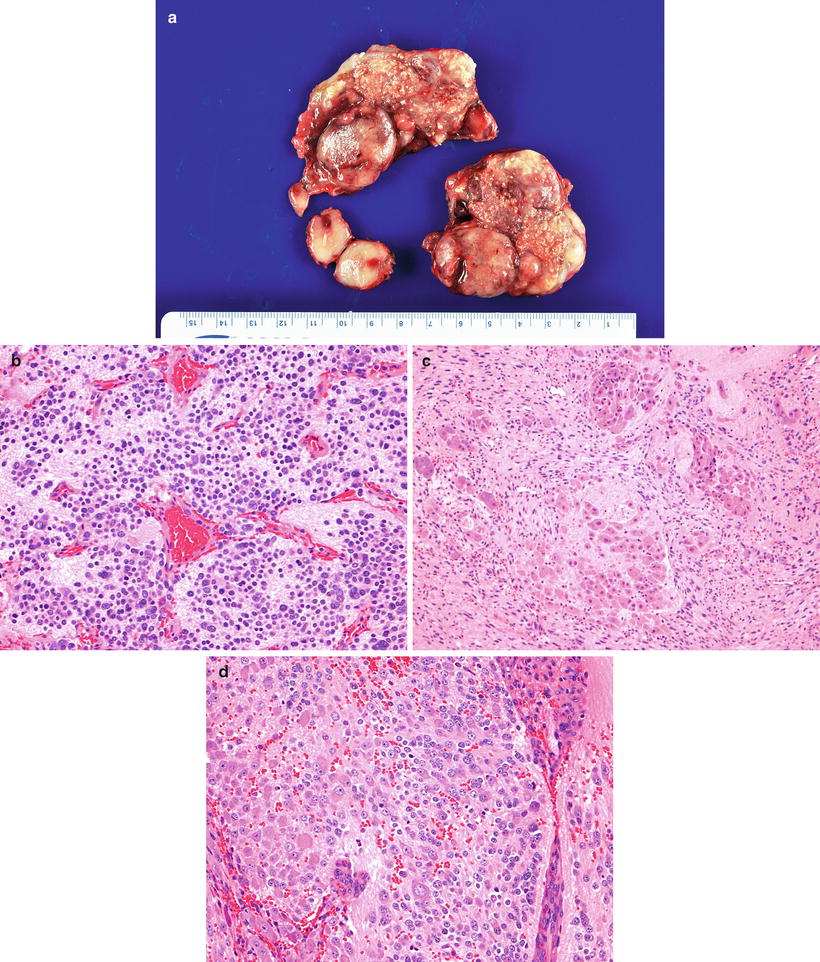
Fig. 12.25
Ganglioneuroblastoma nodular (composite Schwannian stroma-dominant/stroma-rich and stroma-poor) arising in posterior mediastinum. (a) Cut section shows a nodular appearance with areas of stippled calcification, nodules with a light tan appearance with foci of hemorrhage and nodules with a fibrous appearance. The lesion contains three different histological patterns: (b) Poorly differentiated NEUROBLASTOMA (Schwannian stroma-poor) with low MKI (H-E20X). (c) Intermixed ganglioneuroblastoma (Schwannian stroma-rich) with extensive Schwannian stroma development and pockets of naked neuropil containing tumor cells at various stages of neuronal differentiation (H-E10X). (d) Neuroblastoma (Schwannian stroma-poor) differentiating subtype containing more than 5 % of tumor cells showing neuroblastic differentiation (H-E20X)
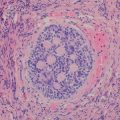
Ganglioneuroma (Schwannian stroma-dominant) is a benign tumor separated into maturing and mature subtypes. The maturing subtype is composed of predominantly ganglioneuromatous Schwann cell stroma with scattered collections of mature/maturing ganglion cells and fully mature ganglion cells. The mature subtype contains mature Schwannian stroma and ganglion cells. Neuroblasts are not present (Fig. 12.26).
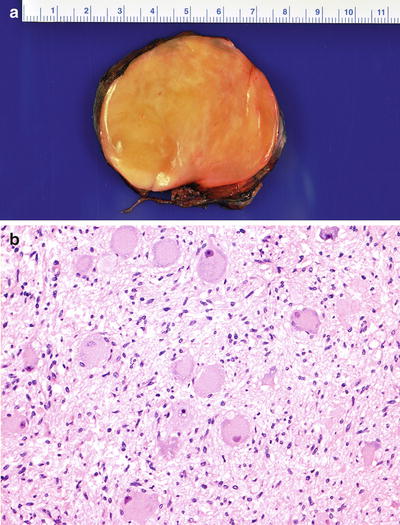
Fig. 12.26
(a) Posterior mediastinal tumor showing a homogeneous light tan color appearance. (b) Ganglioneuroma (Schwannian stroma-dominant) composed of Schwann cells and maturing/mature ganglion cells (H-EX20)
Immunohistochemistry and Other Special Stains
By immunohistochemistry, neuroblastic tumors express neuronal proteins such as neuron-specific enolase (NSE), synaptophysin, Pgp9.5, NB84, and CD56. CD99 is generally negative; positive tumors should be tested for EWS fusions to exclude a Ewing’s sarcoma. More differentiated tumors express chromogranin and neurofilaments. Schwann cell marker expression depends on the relative Schwann cell content.
Molecular Diagnostic Features and Cytogenetics
Hyperdiploid neuroblastoma (with near triploid DNA content) associates with more favorable outcomes, whereas near diploid DNA content predicts poor outcome [58]. Recurrent segmental copy number alterations, mainly deletions of chromosome 1p,3p,11q, and gain of 1q, 2p, and 17q, have been associated with a poor outcome [58]. Loss of 1p36, seen in 25–35 % of neuroblastoma cases [73], has a strong correlation with MYCN amplification and advanced-stage disease, and is associated with an increased risk of disease relapse in localized tumors [73–75]. Allelic loss of 11q, present in 35–45 % of primary tumors [73], rarely occurs with MYCN amplification [76] yet remains associated with other high-risk features; may be the most common deletion event in neuroblastoma [77]. Partial gain of distal 17q, present in up to 50 % of the tumors, is associated with an adverse outcome, particularly in localized tumors without MYCN amplification [78]. Gain of 17q also correlates with a more aggressive phenotype and often with unbalanced translocations with chromosome 1 or 11 [73, 78]. The presence of segmental alterations was the strongest predictor of relapse in neuroblastoma patients in one study [58].
MYCN amplification at chromosome 2p24 is the most important genetic abnormality for risk stratification of neuroblastoma. It occurs in about one fourth of neuroblastoma cases and is associated with advanced-stage disease and rapid tumor progression [79]. Karyotypically, amplification of MYCN manifests as extrachromosomal double minutes or intra-chromosomal homogeneous staining regions. The association of MYCN amplification with poor outcome in patients with otherwise favorable disease patterns, such as localized or 4S disease, underscores its biological importance [15, 79].
Somatic mutations of ALK play a role in both familial [60] and sporadic [80] neuroblastoma. ALK encodes a transmembrane receptor tyrosine (RTK) that is preferentially expressed in the central and peripheral nervous system. Interestingly, the spectrum of somatic and germline ALK mutations varies [58], possibly indicating differences in ALK signaling associated with specific mutations. ALK mutations occur at a high frequency in advanced-stage neuroblastoma [80].
Neurotrophins and their receptors, such as the Trk family, regulate survival, growth, and differentiation of normal and neoplastic neuroblasts and play an important role in the biology and clinical behavior of neuroblastomas. High levels of Trk-A expression in neuroblastoma are associated with a favorable outcome [81], and expression of Trk-C correlates with a favorable prognosis [82, 83]. Truncated Trk-B is predominantly expressed in differentiated tumors, whereas co-expression of full-length Trk-B is associated with MYCN amplification, possibly as an autocrine survival pathway [81, 84].
Prognostic Features
Neuroblastoma is a heterogeneous tumor characterized by a broad spectrum of clinical behaviors ranging from spontaneous regression to fatal outcome despite aggressive therapy. Various clinical and biological factors appear to have prognostic value and have led to risk classifications for patients with newly diagnosed neuroblastoma. Most cooperative groups use a system that combines the patient’s age, tumor stage, and specific biologic variables to derive low, intermediate, and high-risk categories.
The International Neuroblastoma Risk Group (INRG) classification system provides a consensus approach for pretreatment risk stratification [85] and includes stage, age, histologic category (international neuroblastoma classification Table 12.2), grade of tumor differentiation, MYCN status, presence/absence of 11q aberrations, and tumor cell ploidy.
Radiologists involved particularly with clinical trials should be familiar with the 2009 INRG Staging (INRGSS) System [85–87]. As distinguished from the International Neuroblastoma Staging System (INSS), which is a postoperative staging system based on the degree of operative resection (Table 12.3), the new INRGSS, based on clinical criteria and image-defined risk factors (IDRFs) in neuroblastic tumors (Table 12.4), attempts to stage patients prior to operation or other therapies [86]. The presence of these risk factors had been validated to be associated with lower complete resection rate and greater risk of operation-related complications. Imaging modalities that are important include computed tomography (CT) and/or magnetic resonance imaging (MRI), I-123-metaiodobenxylguanidine (MIBG), and technetium-99m-scintigraphy. In the INRGSS based on IDRFs, four stages have been described (Table 12.5).
Table 12.3
The original international neuroblastoma staging system (INSS) tumor stages [Brodeur]
1 | Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically. Nodes attached to and removed with the primary tumor may be positive |
2A | Localized tumor with incomplete gross excision; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically |
2B | Localized tumor with or without complete gross excision, with ipsilateral nonadherent lymph nodes positive for tumor; enlarged contralateral lymph nodes negative microscopically |
3 | Unresectable unilateral tumor infiltrating across the midline (beyond the opposite side of the vertebral column) with or without regional lymph node involvement, or midline tumor with bilateral extension via infiltration (unresectable) or lymph node involvement |
4 | Any primary tumor with dissemination to distant lymph nodes, bone marrow, liver, skin, and/or other organs (except as defined for stage 4S disease) |
4S | Localized primary tumor (as defined for stage 1, 2A, or 2B disease) with disseminated limited to skin, liver, and/or bone marrow (limited to infants younger than 1 year, marrow involvement of less than 10 % of total nucleated cells, and MIBG findings negative in the marrow) |
Multiple body compartments (Ipsilateral tumor extension within two body compartments: (neck-chest, chest-abdomen, abdomen-pelvis) |
Neck |
Tumor encasing carotid and/or vertebral artery and/or internal jugular vein |
Tumor extending to base of skull |
Tumor compressing the trachea |
Cervico-thoracic junction |
Tumor encasing brachial plexus roots |
Tumor encasing subclavian vessels and/or carotid and/or vertebral arteries |
Tumor compressing the trachea |
Thorax |
Tumor encasing the aorta and/or major branches |
Tumor compressing the trachea and/or principal bronchi |
Lower mediastinal tumor, infiltrating the costo-vertebral junction between T9-T12 |
Thoraco-abdominal |
Tumor encasing the aorta and/or vena cava |
Abdomen/pelvis |
Tumor infiltrating the portal hepatis and/or the hepaticoduodenal ligament |
Tumor encasing branches of the superior mesenteric artery at the mesenteric root |
Tumor encasing the origin of the celiac axis, and/or of the superior mesenteric artery |
Tumor invading one or both renal pedicles |
Tumor encasing the aorta and/or vena cava |
Tumor encasing the iliac vessels |
Pelvic tumor crossing the sciatic notch |
Intraspinal tumor extension whatever the location provided that more than one third of the spinal canal in the axial plane is invaded and/or the perimedullary leptomeningeal spaces are not visible and/or the spinal cord signal is abnormal |
Infiltration of adjacent organs/structures: pericardium, diaphragm, kidney, liver, duodenpancreatic block, and mesentery |
Conditions to be recorded, but not considered IDRFs: multifocal primary tumors, pleural effusion, with or without malignant cells, ascites with or without malignant cells
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|