Congenital central nervous system (CNS) tumors represent a distinct group of tumors that differs from familial and genetic CNS tumors. It is accepted that these tumors include those present at birth or diagnosed during the first year of life.
Congenital CNS tumors have been reported to represent between 0.5% and 1.5% of all pediatric brain tumors.1 The fact that many of the congenital CNS tumors often result in intrauterine fetal demise makes the accurate assessment of the true incidence difficult.
The incidence in the United States as reported in the Third National Cancer Survey in 1971 was 14 CNS tumors per 1 million live births per year.2 A similar study from Germany showed an incidence much higher (3.6 per 100,000 births).3
We still have no knowledge or means to identify the cause or causes of malignancies that occur in early life. Fetal and/or maternal exposure to exogenous factors, including ionizing irradiation, drugs, and viruses, may start the biological mechanisms responsible for tumor formation.4 Developmental errors during embryonic and fetal maturation may result in congenital tumors.5 More than 100 years ago, Durante6 and Cohnheim7 proposed the “cell rest” theory that may be adapted to embryonic tumors. These authors believed that more cells are produced than required for the formation of an organ or tissue and that the origins of embryonic tumors rest in developmental errors in these surplus embryonic rudiments. Embryonic tumors developing after infancy are explained by the persistence of cell rests or developmental vestiges.8 Developmentally anomalous tissues (ie, hamartomas and dysgenic gonads) are a source of neoplasms in older children and adults. When any of these developmentally abnormal tissues are present at birth, it is inferred that the cells failed to mature, migrate, or differentiate properly during intrauterine life.
A genetic model of carcinogenesis has been introduced in an attempt to clarify the pathogenesis and behavioral peculiarities of certain embryonic tumors.9 According to this hypothesis, embryonic neoplasms arise as a result of two mutational events in the genome. The first mutation is prezygotic in familial cases and postzygotic in nonfamilial; the second mutation is always postzygotic.
The concept that teratogenesis and oncogenesis have shared mechanisms has been well documented by numerous examples. There is probably simultaneous or sequential cellular and tissue reaction to specific injurious agents. The degree of cytodifferentiation, the metabolic or immunological state of the embryo or fetus, and the length of time of exposure to the agent will determine whether the effect is teratogenic, oncogenic, both, or neither. Many biological, chemical, and physical agents known to be teratogenic to the fetus or embryo are carcinogenic postnatally.10 Alternatively, a teratogenic event during intrauterine life may predispose the fetus to an oncogenic event later in life. This would explain neoplastic transformation occurring in hamartomas, developmental vestiges, heterotopias, and dysgenetic tissues. It is postulated that the anomalous tissues harbor latent oncogenes that, under certain environmental conditions, are activated, resulting in malignant transformation of a tumor.
The type of chromosomal abnormalities appearing in the pediatric brain tumors differs from that found in adult brain tumors. Genetic abnormalities detected at the chromosomal and molecular level have been observed in several fetal brain tumors.11,12 The most common abnormality in medulloblastoma is the partial or total loss of chromosome 17, a monosomy of (17q), which is observed in ~50% of tumors.13 Chromosome 1 deletions (del 1q) have been reported in childhood astrocytomas.14 Studies conducted on fetal brain teratomas have found that a normal 46XY or 46XX karyotype exists for both the patient and tumor.15
An interesting association between monosomy 22, the most frequent chromosomal abnormality in pediatric and adult meningiomas, has been described with primitive neuroectodermal tumor.16 Also, the gene for neurofibromatosis has been mapped to the long arm of chromosome 22. It may be possible to suspect a possible association between neurofibromatosis and CNS tumors sharing a common pathogenetic mechanism in tumorigenesis.17
Large studies on fetal intracranial tumors have provided data about the different histologic subtypes.18,19,20,21 Interestingly, the distribution of the various types of perinatal brain tumors varies in different countries and medical centers. Astrocytoma, medulloblastoma, and choroid plexus papilloma are the major neuroglial perinatal tumors, and teratoma is the leading nonneuroglial tumor. The teratomas are responsible for the largest number of stillbirths, accounting for over one-third to one-half of cases.
Incorporation of electron microscopy and immunohistochemistry techniques is mandatory for identification and classification of brain tumors. The immunohistochemical markers specifically used for evaluation of CNS neoplasms include the glial fibrillary acidic protein (GFAP) and the neurofilament protein (NFP) antibodies.22,23 The GFAP is helpful in distinguishing between glial and nonglial neoplasms and in identifying astrocytic elements. Monoclonal antibodies directed against neuroectoderm-associated antigens include neuron-specific enolase (NSE) and the S-100 protein, which cross-reacts with many types of tissues, both neural and nonneural.24 Tumors such as neuroblastoma, medulloblastoma, and primitive neuroectodermal tumor (PNET) generally exhibit NSE positivity and are focally positive to S-100 protein. Synaptophysin has been found to be a useful marker for identifying these tumors. Alpha-fetoprotein (AFP) and human chorionic gonadotropin (hCG) immunoperoxidase antibodies also aid in the diagnosis of intracranial germ tumors, yolk sac tumor, and choriocarcinoma. For tumors posing a severe diagnostic challenge, one may use electron microscopy.22 Karyotyping is increasingly becoming an aid in detecting certain brain tumors. We will present the pathologic hallmarks of the most common fetal brain tumors.
Intracranial teratoma is the most common intracranial neoplasm diagnosed during the first year of life and the leading cause of tumor-related fetal death.18,19,21 Perinatal intracranial teratomas consist of mature and immature tissues of various types. Most of them arise from midline supratentorial locations, such as the pineal body and third ventricle and suprasellar regions, but occasionally they originate in the cerebral hemispheres or brainstem.25 In ~50% of cases, the teratoma reaches gigantic size, effacing normal brain tissue elements; thus, it destroys any identifiable clue for site of origin.
A classification of intracranial teratomas into three subtypes has been proposed:26
Teratomas replacing the brain tissue (Figure 13–1)
Smaller tumors usually not causing hydrocephaly (Figures 13–2, 13–3, and 13–4)
Teratomas with local extension into the face (Figures 13–5 and 13–6)





Figure 13–6.
Post mortem picture of the fetus in Figure 13–5. Note the macrocephaly and eye protrusion due to extension of the tumor into the orbit. (Courtesy of Ron Rabinovitz, MD, Jerusalem, Israel.)

Hydrocephaly may be the initial finding. In several cases it has been reported that hydrocephaly has progressed rapidly on serial scans prior to the detection of a huge teratoma.15 This observation suggests that early obstruction to the flow of cerebrospinal fluid (CSF) by the midline location of the tumor is followed by effacement of the brain by continued tumor growth. Neonates, having small lesions, may show evidence of hydrocephaly after birth caused by aqueductal stenosis.
Despite recent advances in diagnosis and therapy, with fetal and neonatal intracranial teratomas, stillbirth or perinatal death occurs in most cases.27,28,29 In some cases, dystocia may happen.30
An extremely rare congenital condition, fetus in fetu has been described as presenting as a mass. The mass may be present intracranially (Figure 13–7), intra-abdominally, retroperitoneally, or in the scrotum. The exact relationship between teratoma and fetus in fetu is controversial.31,32,33,34,35,36 Some feel that this might in fact be two edges of the same entity, namely, a teratoma.32 However, it has been stated that to distinguish between fetus in fetu and teratoma, the vertebral column must be present.37
Figure 13–7.
A very rare case of a true intracranial fetus-in-fetu that presented at 17 weeks with an intracranial mass. Upon closer inspection, the mass had the sonographic appearance of a fetus. (A) Transabdominal scan at 17 weeks showing intracranial mass (arrow). Patient elected to terminate the pregnancy. (B) Picture of the gross specimen showing the well-formed fetus (arrow) within the cranial cavity pushing the brain to one side. (C) Histology demonstated a well-formed fetal vertebral column. (Courtesy of Prof. A. lanniruberto, Italy.)

Astrocytoma is the leading neuroglial tumor of infancy. In the fetus, it is usually supratentorial. The cerebral hemisphere is the most common primary site (two-thirds of cases). Those arising from the cerebral hemispheres are frequently large, may involve more than one lobe, and occupy an extensive part of the brain (Figure 13–8). Hydrocephaly is the main presenting feature in both fetus and neonate, regardless of site of origin. Almost half of perinatal astrocytomas are malignant, and some have been called anaplastic astrocytomas.
Figure 13–8.
Four views of the brain of a fetus at 34 postmenstrual weeks having a tumor of the brain. (A) Midcoronal–3, almost occipital–1, section showing the dilated right and left lateral ventricles, the thin and dangling hyperechoic choroid plexus, and the extremely thin posterior fossa (arrow). (B) Median section showing the extent of the tumor in the midline, above and behind which the dilated ventricles are seen. (C) Midcoronal–1 section. (D) Horizontal section. After the diagnosis of the space-occupying lesion in the brain, causing severe obstructive hydrocephaly, it was decided that the patient’s labor would be induced. Cephalocentesis, to decrease the size of the head by draining cerebrospinal fluid (csf), was performed. (E) The needle is seen in the fluid at a depth of 6.5 cm. The head circumference decreased from 40 cm to enable vaginal birth, which was achieved after inducing labor. (F) The stillborn neonate was examined by the pathologist. The arrow indicates the third ventricle, and the double arrow indicates the longitudinal sulcus. Histologic examination of the tumor revealed a glioblastoma. (Courtesy of Susan M. Staugaitis, MD, PhD; Departments of Neurosciences and Anatomic Pathology, Cleveland Clinic.)

Originating from the glia, glioblastoma multiforme (GBM) this is an extremely rare tumor in the fetal brain. Its prenatal diagnosis is possible and was reported to occur usually after 25 weeks.38 Fetal glioblastomas are characteristically fast-growing intracranial tumors presenting as polyhydramnios and hydrocephaly and a fast-growing echogenic, vascularized brain lesion (see Figure 13–8). Most cases of GBM occur supratentorially. The prognosis is almost uniformly grim. All neonates in the published literature succumbed at birth or shortly thereafter.39,40,41,42,43,44,45
PNET occurs in several locations, including the cerebellum, cerebral hemispheres, pineal body, brainstem, spinal cord, olfactory nerve, and retina24 (Figure 13–9). Microscopically, PNETs are represented by groups of small, poorly differentiated, darkly stained cells. They occur primarily in the pediatric age group and are characterized by very aggressive behavior, regardless of their primary site or histologic components. This type of tumor metastasizes widely within the CSF pathways and seeds the meninges of the brain and spinal cord.22
Figure 13–9.
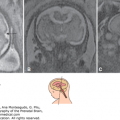
Primitive neuroectodermal tumor. (A) Fetal brain MRI at 34 weeks of gestation shows the mass involving the brain parenchyma (white arrow) and extending into the lateral ventricle (black arrow). Note the presence of marked ventriculomegaly. (B) Brain MRI at 5 years of age shows complete remission following surgery. (Courtesy of Daniela Prayer, Vienna, Austria.)

Medulloblastoma, also called PNET of the cerebellum, is the leading infratentorial tumor. It is associated with a high frequency of stillbirth, hydrocephaly, and congenital defects.46 Anomalies described to appear in conjunction with this tumor include cleft palate, omphalocele, malrotation of the intestine, imperforate anus, and bladder exstrophy.46 A tendency toward a familial occurrence of this tumor has been reported.47 An association with rhabdoid tumor of the kidney has also been reported, suggesting that the neural crest is the common denominator.48
Choroid plexus papilloma is a benign tumor composed of epithelial cells that line the ventricular choroid plexuses.49 The incidence is inversely correlated with age, and ~50% of cases in patients in the pediatric age group are detected during the first year of life.50 These neoplasms rank third in frequency of congenital CNS tumors. The main presentation is rapidly developing hydrocephaly caused by the growing papilloma into the lateral ventricles, sometimes into the third or fourth ventricle. This overgrowth produces a large, space-occupying nodular lesion that is readily observed on imaging studies (Figure 13–10). The tumor may produce large amounts of CSF, causing hydrocephaly. The prognosis of patients having choroid plexus papilloma is comparatively good and is the most favorable of all newborn brain tumors.51 It is important to diagnose choroid plexus papilloma preoperatively, as surgical removal is usually curative.
Figure 13–10.
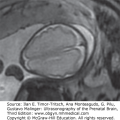
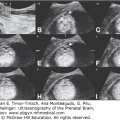
Choroid plexus papilloma diagnosed at 39 weeks, 6 days of gestation. Ventriculomegaly was observed during a routine ultrasound (US) examination for weight estimation. The child underwent complete resection of the tumor with normal postoperative follow-up. (A) Axial transventricular plane shows ventriculomegaly (arrows). The measured lateral ventricle width was 16 mm. (B) Axial transthalamic plane shows a midline, well-defined, echogenic mass (arrows). (C) Coronal transthalamic plane at the level of the foramen of Monro shows the mass (arrows) in the third ventricle, causing separation of the thalami (T). Note dilation of the frontal horns of the lateral ventricles (LV). (D) Median plane shows the tumor completely filling the third ventricle (arrows). CSP, cavum septi pellucidi; CV, cavum verga; arrowheads, corpus callosum. (E) Color Doppler at the same plane as in Figure 13–9C shows the vascularity of the tumor mass, thus making it possible to differentiate between hemorrhage and tumor. (Courtesy: Gustavo Malinger.)
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


