Etiology, Prevalence, and Epidemiology
Idiopathic pulmonary fibrosis (IPF) has been defined as “a specific form of chronic fibrosing interstitial pneumonia limited to the lung and associated with the histologic and/or CT appearance of usual interstitial pneumonia (UIP).” IPF is the most common idiopathic interstitial lung disease. Data from the late 1980s estimated a prevalence of 20 per 100,000 for men and 13 per 100,000 for women, and an incidence of 11 per 100,000 per year in men and 7 per 100,000 per year in women. However, more recent data has shown that prevalence and incidence of IPF have increased, with a prevalence of 42.7 to 63 per 100,000 and an incidence of 16.3 to 17.4 per 100,000 per year, which may reflect the average increased life expectancy in that span of time. Most patients with IPF are older than 50 years, and approximately 80% are older than 65 years. Indeed, age older than 75 years has been shown to have an extremely high positive predictive value in IPF diagnosis. IPF has been reported worldwide in rural and urban settings. IPF is likely more common in Caucasians, although this has not been definitively proven. IPF is more common in smokers; the odds ratio of IPF in smokers compared with lifelong nonsmokers in various studies ranges from 1.6 to 2.9. Occasionally, clusters of IPF may occur in families. Although the prevalence of familial IPF is unknown, it has been estimated to account for 0.5% to 2.2% of patients with IPF.
Clinical Presentation
Symptoms are nonspecific and include progressive dyspnea, nonproductive cough, weight loss, and fatigue. Dyspnea is usually the main symptom and typically is present for more than 6 months before initial diagnosis. Diagnosis is delayed by more than 1 year in up to half of patients. Finger clubbing occurs in 25% to 50% of patients. Auscultation typically reveals late inspiratory crackles (so-called Velcro rales). Cyanosis and signs of pulmonary hypertension and cor pulmonale are late manifestations.
The clinical course of IPF is typically one of relentless progressive increase in the extent of fibrosis and severity of clinical manifestations. The course of individual patients varies, however, and some patients remain stable for extended periods without treatment. Acute deterioration with an abrupt and unexpected worsening of symptoms may occur secondary to infection, pulmonary embolism, pneumothorax, or heart failure. Often, no identifiable cause for the acute decline is identified, however, and these episodes are called “acute exacerbation” or “accelerated phase” of IPF.
Acute exacerbations of IPF are defined as an acute, clinically significant respiratory deterioration characterized by new widespread alveolar abnormality and are considerably more common than previously recognized. In one retrospective study of 168 patients with IPF, 21% of patients died over a median period of 76 weeks, and 47% of these deaths followed an acute deterioration in the patient’s clinical status. In a second study of 147 patients with biopsy-proven IPF, the 1-year frequency of acute exacerbation was 8.5%, and the 2-year frequency was 9.6%. As pointed out by the authors, however, the actual incidence of acute exacerbation could have been underestimated in their study because some of the patients were lost to follow-up and because the study included only patients with biopsy-proven IPF. In an autopsy series of 25 patients, 15 (60%) deaths in patients with IPF or nonspecific interstitial pneumonia (NSIP) associated with connective tissue disease were due to diffuse alveolar damage and consistent with acute exacerbation. The reported mortality rates from acute exacerbation range from 20% to 86%. Diagnostic criteria for acute exacerbation of IPF have recently been revised and include:
- 1.
Previous or concurrent diagnosis of IPF
- 2.
Acute worsening or development of dyspnea typically of less than 1 month in duration
- 3.
CT findings of bilateral ground-glass opacities and/or consolidation superimposed on a background pattern consistent with usual IPF
- 4.
Deterioration not fully explained by cardiac failure or fluid overload
Pathophysiology
Pathology
It is currently believed that several components are involved in the pathogenesis of IPF, including repetitive lung injury, inflammation, exaggerated deposition of collagen and extracellular matrix, recruitment and proliferation of fibroblasts, inappropriate wound healing response, and excessive angiogenesis. Epithelial injury and activation of fibroblast foci are currently considered the main early events that trigger a cascade of changes that eventually lead to extensive fibrosis.
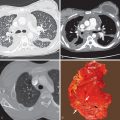
IPF by definition is idiopathic UIP. The key histologic features of UIP are temporal and geographic heterogeneity, presence of fibroblastic foci, and predilection for the periphery of the lobule and the subpleural regions ( Fig. 27.1 ). Additional features include smooth muscle hypertrophy, excessive collagen and extracellular matrix, destroyed alveolar architecture, traction bronchiectasis and bronchiolectasis, and honeycombing. Temporal heterogeneity refers to the presence of active inflammation, scattered fibroblastic foci, and chronic scarring (i.e., findings ranging from relatively acute disease to established fibrosis). This is distinct from other interstitial pneumonias, such as NSIP, in which the changes seem to have occurred over a relatively narrow time span. Geographic heterogeneity refers to the presence of alternating areas of normal lung, interstitial inflammation, fibrosis, and honeycombing in different regions of the same tissue section and sometimes in a single lobule. Fibroblast foci are aggregates of proliferating fibroblasts and myofibroblasts and represent microscopic zones of acute lung injury on a background of chronic scarring (see Fig. 27.1 ). Fibroblastic foci are essentially areas of immature fibrosis that signify the active, advancing edge of lung remodeling.

Areas of honeycombing consist of cystic fibrotic airspaces, which frequently are lined by bronchiolar epithelium and filled with mucin. Honeycombing usually represents markedly dilated transitional airways (respiratory bronchioles and alveolar ducts) and typically is associated with severe fibrosis and destruction of lung architecture. The honeycomb cysts usually range from 2 to 20 mm in diameter and are separated by a variable amount of fibrous tissue ( Fig. 27.2 ). Although interstitial inflammation commonly is seen in UIP, it is typically mild and patchy. Dystrophic calcification and osseous metaplasia occur occasionally.

In patients who undergo biopsy during an accelerated phase of their illness (acute exacerbation of IPF), the specimens show a prominent organizing pneumonia or diffuse alveolar damage pattern superimposed on the UIP pattern. Patients with organizing pneumonia have a better prognosis than patients with diffuse alveolar damage.
Most patients with a histologic pattern of UIP have IPF, and by definition all patients with IPF have a histologic pattern of UIP. A histologic pattern of UIP is not diagnostic of IPF, however. A UIP pattern also may be seen in patients with connective tissue diseases, asbestosis, chronic hypersensitivity pneumonitis, certain drug-induced lung diseases, and familial IPF. These conditions must be excluded before making a diagnosis of IPF.
Lung Function
IPF results in restrictive lung function (decreased total lung capacity and vital capacity) and impaired gas transfer as assessed by the carbon monoxide diffusing capacity. Most patients have normal or even increased expiratory flow rates when related to absolute lung volume. A few patients have evidence of airflow obstruction manifested by reduction in the maximal midexpiratory flow rate and forced expiratory volume in 1 second relative to the reduction in vital capacity, probably reflecting the effects of cigarette smoking. Indices of airflow obstruction correlate closely with the presence of emphysema as determined by high-resolution CT. Hypoxemia is common and is caused mainly by ventilation-perfusion inequality; however, in about 20% of cases, it can be attributed to a diffusion defect.
Several studies have shown that the extent of abnormalities of high-resolution CT in patients with IPF correlates with severity of functional impairment. Among the functional parameters, carbon monoxide diffusing capacity correlates best with extent of disease on CT.
Manifestations of the Disease
Radiography
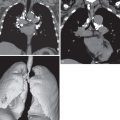
The radiographic findings of UIP usually consist of symmetric, basal, small- to medium-sized irregular linear opacities resulting in a reticular pattern ( Fig. 27.3 ). Although these opacities may be diffuse throughout both lungs, in 50% to 80% of cases they involve the lower lung zones predominantly or exclusively; in 60% a predominant peripheral distribution is apparent. As the disease progresses the abnormalities become more diffuse and assume a coarser reticular or reticulonodular pattern associated with progressive loss of volume ( Fig. 27.4 ). Advanced disease is characterized by the presence of honeycomb cysts measuring 0.5 to 1 cm in diameter ( Fig. 27.5 ). Evidence of pleural disease (effusion, pneumothorax, or diffuse thickening) is uncommon. Decreased lung volumes are radiographically evident at initial evaluation in 50% to 60% of cases. The lung volumes decrease progressively over time. Most patients with IPF are smokers. In patients with associated emphysema the lung volumes may be normal or increased.



Virtually all patients with IPF have an abnormal chest radiograph at the time of presentation. Basal reticular opacities are often evident on previous chest radiographs for several years before the development of symptoms. A normal chest radiograph cannot be used, however, to exclude microscopic evidence of UIP.
Computed Tomography
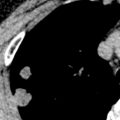
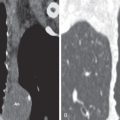
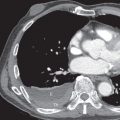
UIP is characterized on high-resolution CT by the presence of patchy irregular intralobular lines resulting in a reticular pattern involving mainly the subpleural regions and the lower lung zones ( Fig. 27.6 ). The reticular pattern is associated with architectural distortion; dilation of bronchi and bronchioles (traction bronchiectasis and bronchiolectasis); and irregular pleural, vascular, and bronchial interfaces ( Fig. 27.7 ). The dilated bronchi and bronchioles in areas of fibrosis typically have a beaded appearance. Air-containing cysts lining up in rows or stacks in the subpleural lung measuring 2 to 20 mm in diameter (honeycombing) are seen in approximately 80% to 90% of patients ( Figs. 27.8 and 27.9 ). Honeycombing is the most specific sign of UIP on CT.




A predominantly subpleural distribution of the reticular pattern and honeycombing is evident on high-resolution CT in nearly all patients. In approximately 80% of patients, the fibrosis is most severe in the lower lung zones; in about 15%, all zones are involved to a similar degree; and in 5%, mainly the upper lung zones are affected. Although the reticulation and honeycombing in UIP tend to be most severe in the lower lung zones, in most patients the reticulation involves all lung zones. The geographic heterogeneity that is described on histologic specimens of UIP also is appreciated on high-resolution CT and refers to the presence of fibrosis (reticulation and honeycombing) alternating with areas of normal lung parenchyma (patchy distribution) (see Fig. 27.9 ).
Ground-glass opacities are present in most patients with UIP. The ground-glass opacities in these patients are usually mild, however, and associated with evidence of fibrosis ( Fig. 27.10 ). Ground-glass opacities away from areas of fibrosis are present in approximately 25% of patients and are typically less extensive than the reticulation. Ground-glass opacities in UIP may reflect the presence of active inflammation or microscopic fibrosis ( Fig. 27.11 ). Ground-glass opacities resulting from microscopic fibrosis usually are associated with reticulation, traction bronchiectasis, or bronchiolectasis (see Fig. 27.10 ). Ground-glass opacities not associated with fibrosis correlate with interstitial inflammation. On serial CT scans in patients with UIP, the areas of ground-glass opacity may regress, but more commonly they progress to reticulation and honeycombing.


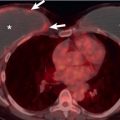
Emphysema is evident on CT in approximately 30% of patients with UIP, with a few localized areas of decreased attenuation and vascularity (mosaic attenuation) in 4% to 43%, small areas of consolidation in 3%, and a few centrilobular nodules in 2% to 15%. Not uncommonly, fine linear or small nodular foci of ossification are seen within areas of fibrosis as a result of diffuse or dendriform pulmonary ossification ( Fig. 27.12 ). Also, it is important to keep in mind that patients with IPF have an increased prevalence of pulmonary carcinoma, and careful inspection of the lungs for focal nodules or slowly growing lung lesions is mandatory.

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


