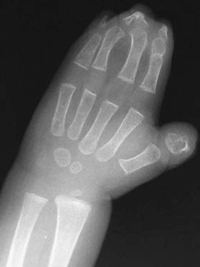
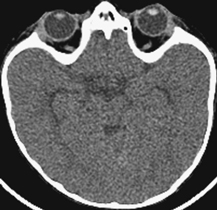
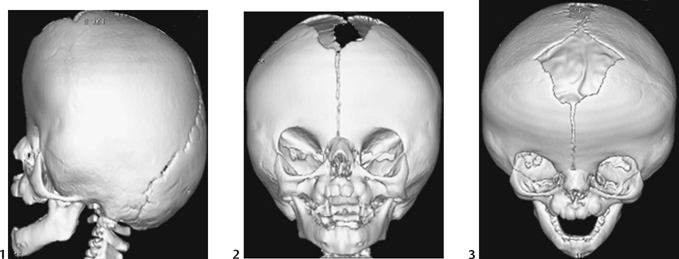
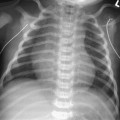
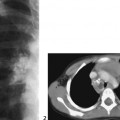
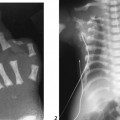
CASE 115 A 16-month-old child presents with hypertelorism, an elongated, flattened forehead, and syndactyly of hands and feet. Figure 115A Figure 115B Figure 115C Radiograph of the left hand (Fig. 115A) demonstrates the characteristic “mitten hand” deformity, with hypoplasia of the first, third, and fourth metacarpals, abnormally short and wide first proximal “delta” phalanx, and symphalangism (i.e., syndactyly/ankylosis of the finger or toe joints) of second through fourth rays, with soft tissue syndactyly of the second through fifth rays. The right hand demonstrated an identical appearance. The anterior fontanelle is widely patent. There is synostosis of the coronal sutures, hypertelorism, and flattening and downward slanting of the shallow bony orbits (Figs. 115B, and 115C). Apert syndrome (acrocephalosyndactyly type 1) Many syndromes and conditions include abnormalities of both the craniofacial structures and the limbs. Patients with syndactyly, distal limb abnormalities, and craniosynotosis most often suffer from acrocephalosyndactyly. Of the specific entities within this grouping, Apert syndrome is the most common. The association between syndactyly and craniosynostosis was first described by Apert in 1906. Apert syndrome shows autosomal dominant heritance, but nearly all cases result from new gene mutations. The incidence of Apert syndrome in the general population is ~1 in 160,000 live births. Apert syndrome appears to be caused by either of two distinct point mutations in fibroblast growth factor receptor 2 (FGFR2). Close phenotypic mimics are Crouzon syndrome and Pfeiffer syndrome, both of which are also caused by mutations in FGFR2. Apert syndrome may be diagnosed prenatally and presents clinically at birth. Cranial findings seen in affected infants include a flattened, elongated forehead, turribrachycephaly, hypertelorism, exophthalmos, a bulbous nose, and a flattened nasal bridge. Nasopharyngeal obstruction is seen in 50%, due to midface hypoplasia. The orbits show downward slanting. Most patients demonstrate at least mild prognathism.
Clinical Presentation
Radiologic Findings
Diagnosis
Differential Diagnosis
Discussion
Background
Etiology
Clinical Findings
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


