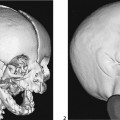
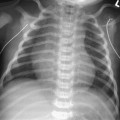
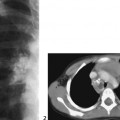
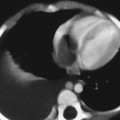
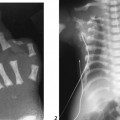
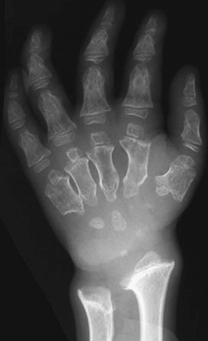
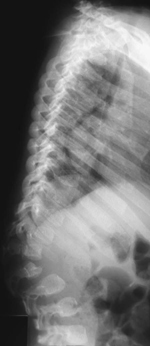
CASE 118 A young child presents with mild kyphosis, symmetrical short stature, coarsened facial features, and developmental delay. Figure 118A Figure 118B Figure 118C Frontal radiograph (Fig. 118A) of the hand demonstrates shortened metacarpals and phalanges. There is marked proximal tapering of metacarpals and distal phalangeal tapering. The distal radial and ulnar metaphyses are widened and irregular. All epiphyses demonstrate a bizarre, irregular appearance. Lateral view of the chest and spine (Fig. 118B) demonstrates broad, “oar”-shaped ribs, short and thick clavicles, rounded scapulae, and osteopenia. There is a gibbus (abnormal, abrupt kyphosis) deformity at the thoracolumbar junction, due to a pronounced anteroinferior beak at L2. Note the appearance of the gibbus with anterosuperior hypoplasia of L2 on sagittal T1-weighted MRI (Fig. 118C). Figure 118D Figure 118E Frontal radiograph of the pelvis shows abnormally steep acetabular roofs, flared, constricted iliac wings, and moderate bilateral coxa valga (Fig. 118D). Parasagittal and midline sagittal T1-weighted MR image of the brain demonstrate frontal bossing and abnormal soft tissue posterior to the dens, causing mild extradural impression on the thecal sac and cervical cord at C1 (Fig. 118E). Hypointense rounded foci are seen within the corpus callosum. The sella is J-shaped. Axial T2-weighted image (Fig. 118F) shows prominent perivascular Virchow-Robin spaces and delayed white matter myelination. Figure 118F Hurler syndrome (mucopolysaccharidosis IH; MPS-IH) Hurler syndrome (MPS-IH) is but one of a group of phenotypically related conditions, often collectively termed mucopolysaccharidoses, that are caused by specific lysosomal enzymatic deficiencies. The enzymatic deficiencies render the lysosomes incapable of catabolizing particular subregions of complex intracellular glycosaminoglycans, resulting in intracellular and extracellular accumulation of these substances. The various entities in this broad disease category result in the intralysosomal accumulation of dermatan sulfate, chondroitin sulfate, heparan sulfate, and keratan sulfate, either specifically or in combination (Table 118A). Phenotypically, most of the entities in this category produce radiographically and clinically similar patterns, and are best diagnosed definitively via urinalysis and cultured leukocyte or fibroblast enzymatic assays. With the exception of Hunter syndrome, all demonstrate autosomal recessive inheritance. The mucopolysaccharidoses produce a variable radiographic constellation of characteristic findings termed dysostosis multiplex. It is important to remember that this is a descriptive and generic term, not a diagnosis in and of itself. Dysostosis multiplex consists of variable proximal metacarpal tapering, anterior vertebral beaking, acetabular roof hypoplasia, diploic thickening, thoracolumbar gibbus formation, broadening of ribs, and short stature. The separate entities differ in the presence or absence of neurodevelopmental delay, severity of skeletal deformities, and associated extraskeletal comorbidities.
Clinical Presentation
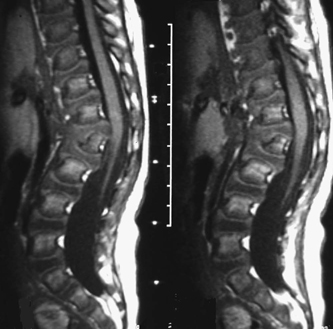
Radiologic Findings
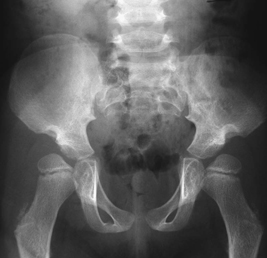
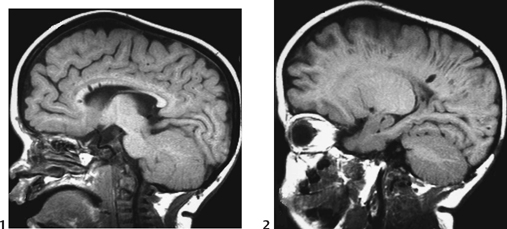
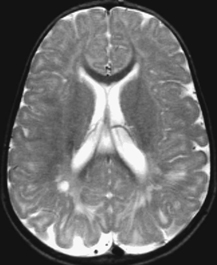
Diagnosis
Differential Diagnosis
Discussion
Background
| Type | Common Eponym | Enzymatic Defect |
| IH | Hurler syndrome | a-L-iduronidase |
| IHS | Hurler-Scheie syndrome | a-L-iduronidase |
| IS | Scheie syndrome | a-L-iduronidase |
| II | Hunter syndrome | Iduronidate sulfatase |