7 Neurocutaneous
7.1 Introduction
A variety of heterogeneous syndromes and syndromic associations produce findings in the central nervous system and the skin, and often elsewhere in the body. These entities are known as neurocutaneous syndromes, or phakomatoses. Some of these syndromes are commonly encountered in a pediatric neuroradiology setting (Table 7-1), while others are more rare (Table 7-2). The genetics of some phakomatoses are well described and understood, while others have no specific genetic foundation that has yet been described (e.g., the posterior fossa malformations–hemangiomas–arterial anomalies–cardiac defects–eye abnormalities–sternal cleft and supraumbilical raphe [PHACES] syndrome). Notwithstanding this, familiarity with the phakomatoses, in particular the more commonly encountered entities among this group of syndromes, can aid in their recognition and appropriate treatment. Accurately recognizing a phakomatosis can prevent its misdiagnosis and can help determine when family genetic evaluation and counseling may be beneficial. Although the genetic causes of many neurocutaneous syndromes have been identified, the family history for such a disorder will often be negative and cases of the syndrome will be the result of new sporadic mutations.
Syndrome | Comment |
Cowden | Lhermitte–Duclos disease, thyroid and breast cancers |
Posterior fossa malformations–hemangiomas–arterial anomalies–cardiac defects–eye abnormalities–sternal cleft and supraumbilical raphe syndrome (PHACES) | Hemangiomas, posterior fossa abnormalities |
Hereditary hemorrhagic telangiectasia | Arteriovenous malformations, and may present with brain abscess due to pulmonary shunt |
Neurocutaneous melanosis | Leptomeningeal melanin deposits (T1 shortening) |
McCune–Albright syndrome | Café au lait spots, precocious puberty, polyostotic fibrous dysplasia |
7.2 Tuberous Sclerosis Complex
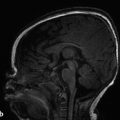
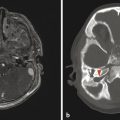
Tuberous sclerosis complex (TSC), or Bourneville disease, is a neurocutaneous syndrome with characteristic manifestations in the brain, eyes, and skin, as well as the heart, lung, and kidneys. 1 The most common presenting symptom is seizures, present in approximately 80% of patients with TSC. Defects in two genes, TSC-1 and TSC-2, which encode the proteins tuberin and hamartin, respectively, are responsible for TSC. Tuberin and hamartin form the TSC dimer, which inhibits the mammalian target of rapamycin (mTOR) pathway. Defects in either TSC-1 or TSC-2 result in dysfunction of the dimer and unregulated (or underregulated) mTOR activity. The findings characteristic of this in the central nervous system (CNS) include eponymous cortical tubers, which the neurologist Désiré-Magloire Bournville, after whom the disease was named, thought resembled potato spuds. Given the firmness of the tubers, the name “tuberous sclerosis” emerged (literally, “hard potato”). The tubers in tuberous sclerosis complex are areas of cortical thickening and gyral expansion, producing abnormal subcortical T2/fluid-attenuated inversion recovery (FLAIR) signals (Fig. 7.1). The signal abnormality of the lesions tapers as they extend inward to the lateral margin of the lateral ventricles, which was the embryologic location of the germinal matrix. Historically, the cortical abnormalities in TSC have been referred to as “tubers” in the radiology literature, and the subcortical linear extensions of these abnormalities toward the location of the germinal matrix have been described as radial migration lines. In fact, the tubers and the radial migration lines are not two separate entities, and the histologic pattern is that of cortical dysplasia (in particular, focal cortical dysplasia [FCD] type IIb) (Fig. 7.1). The areas of dysplasia can at times have calcifications, and there can occasionally be cystic changes. It is worth noting that in the newborn, these areas of dysplasia are best depicted on T1W imaging (Fig. 7.2), and are typically difficult to identify on T2W and FLAIR imaging because of the high water content of the surrounding unmyelinated brain. From approximately 3 to 24 months of patient age, the areas of dysplasia can be difficult to see on either T1W or T2W/FLAIR imaging because of ongoing myelination.
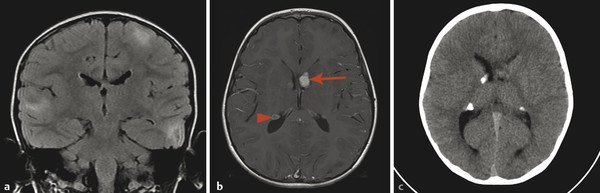
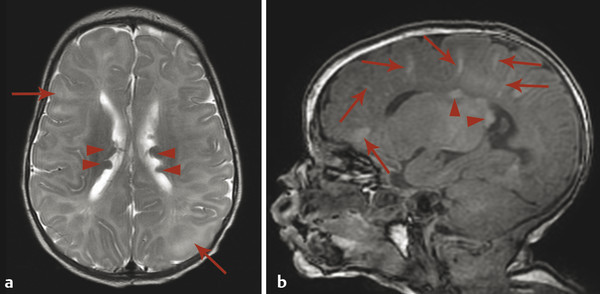
Another characteristic feature of TSC is subependymal hamartomatous nodules along the margins of the lateral ventricles (Fig. 7.1). These nodules frequently calcify, which can be best depicted on computed tomographic scans; however, magnetic resonance imaging (MRI) (and particularly-susceptibility weighted imaging [SWI]) can often give clues about their calcification. The subependymal nodules will often enhance after the injection of gadolinium because there is no intact blood–brain barrier. Large (>10 mm) or enlarging nodules are low-grade neoplasms known as subependymal giant-cell astrocytomas (SEGAs), but even small nodules probably fall within this pathophysiologic spectrum. The most common place for SEGAs is along the lateral margin of the lateral ventricles at the level of the foramina of Monro (Fig. 7.1). It is important to note that subependymal nodules and SEGAs are not “tubers.”
Although traditionally treated with surgical resection, SEGAs have now been shown to involute on therapy with mTOR inhibitors, such as rapamycin or everolimus. These inhibitors reduce the size of SEGAs to a point at which they have decreased mass effect, which can facilitate their elective surgical resection or allow their observation without surgery. 2
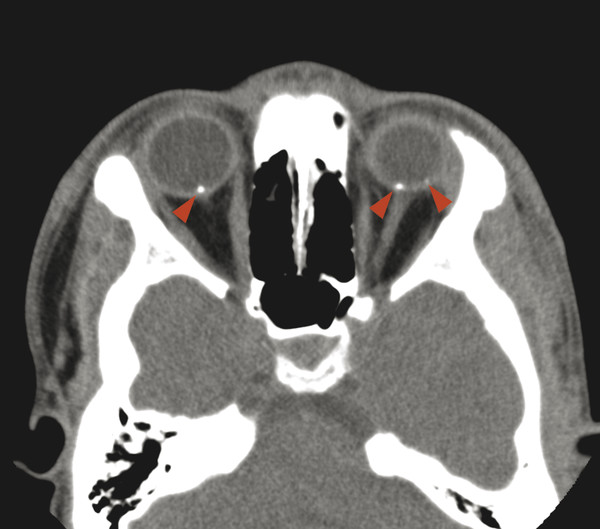
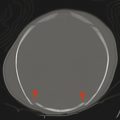
Cerebellar findings are less common than cerebral findings in TSC, but they do occur. Attention to the globes, especially on computed tomographic scans and in SWI, can show signs of retinal hamartomas (Fig. 7.3). Although the latter is an important observation, dilated funduscopic examination is more sensitive, and a majority of retinal hamartomas are not visible on imaging.
There are no associated spinal findings in TSC, but if spinal imaging is done, attention to the imaged portions of the kidneys is warranted to look for signs of angiomyolipomas. The characteristic skin lesions in TSC include facial angiofibromas (adenoma sebaceum), ash-leaf spots, and shagreen patches. Adolescent and young adult females with tuberous sclerosis are at increased risk for developing a lymphangioleiomyomatosis-like interstitial lung disease with thin-walled cystic changes. Infants with TSC may have cardiac rhabdomyomas, which may even be detected in utero, but these tend to spontaneously regress.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


