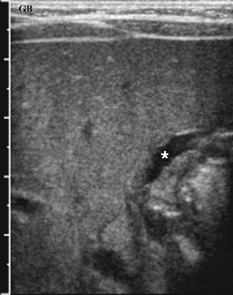
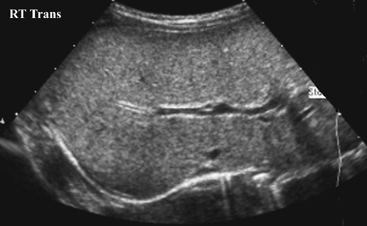
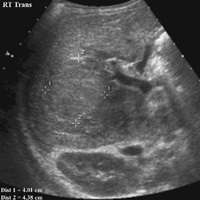
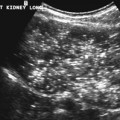
CASE 70 A 2-week-old neonate presents with jaundice (conjugated hyperbilirubinemia). Figure 70A Figure 70B Figure 70C Small size of gallbladder (Fig. 70A, seen as *) and area of increased echogenicity at porta hepatis but no dilatation of bile ducts on ultrasonography (Fig. 70B). Hepatic iminodiacetic acid (HIDA) scan shows adequate hepatocyte uptake but no excretion in bowel by 24 hours (Fig. 70C). Biliary atresia Neonatal cholestasis may result from a wide variety of etiologies. In up to 80% of the cases, the primary differential is idiopathic neonatal hepatitis versus biliary atresia. Biliary atresia is a condition that is initiated by the disruption of the normal extrahepatic biliary system. Biliary atresia is characterized by the destruction or absence of portions of the biliary system at a point(s) between the duodenum and the liver. Progressive damage of extra- and intrahepatic bile ducts secondary to inflammation may result in progressive sclerosis of biliary tissue, biliary cirrhosis, and eventual liver failure. Biliary atresia is seen in 1 of 10,000 to 15,000 births in the United States with higher incidence in the Asian population. A slight female predominance is noted. Two distinct forms of biliary atresia are known: the fetal/embryonic form and the postnatal form. The fetal form presents in the first 2 weeks of life and accounts for 10 to 35% of cases. In this case, the ducts are discontinuous at birth, and 10 to 30% of affected children have associated congenital defects, including situs inversus, polysplenia, malrotation, intestinal atresia, and cardiac anomalies. The postnatal form presents in children 2 to 8 weeks of age and accounts for 65 to 90% of cases. These children may experience a short jaundice-free interval, and, unlike the fetal form, this form is not associated with congenital anomalies. Although exact numbers may vary, overall survival of children post-surgical treatment for biliary atresia is —60% at 5 years of age and 35% at 10 years of age. Figure 70D Ultrasound of regenerative liver nodule in a child with cirrhosis. This patient presented with hematemesis following failed Kasai operation for biliary atresia associated with malrotation and cat-eye syndrome. Its precise etiology is unknown. It is thought to result from inflammation of the hepatobiliary system. Obstruction to flow leads to secondary biliary cirrhosis. Many hypotheses have been proposed for its pathogenesis: antenatal infection with Reovirus type 3, biliary epithelial sloughing due to reflux of pancreatic juice because of abnormal connection between distal bile duct and pancreatic duct, and autoimmune destruction of the biliary epithelium.
Clinical Presentation

Radiologic Findings
Diagnosis
Differential Diagnosis
Discussion
Background
Etiology
Clinical Findings
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


