Abstract
Anomalies of the hands are often difficult to precisely diagnose. Hand malformations range from subtle deformities such as isolated fifth finger clinodactyly to abnormalities of each phalange, including the thumb. There are multiple thumb abnormalities that are associated with hundreds of different disorders. Thumbs can be aplastic, hypoplastic, triphalangeal (three phalange bones instead of two), or broad or have abnormal angulation. Any of these abnormalities can occur in isolation, or they can be associated with a multitude of aneuploidies or genetic syndromes. Detection of abnormalities in any organ system warrants evaluation of the fetal hand, including the thumb, and detection of hand abnormalities, including those of the thumb, warrant detailed interrogation of all organ systems, including the skeleton.
Keywords
hands, thumbs, Holt-Oram syndrome, Fanconi Anemia, Apert syndrome
Introduction
Routine second-trimester ultrasound (US) examination typically involves only a cursory evaluation of the extremities, documenting the presence or absence of the arms and legs; however, comprehensive evaluation of the fetal extremities often yields critical information. Anomalies of the hands are difficult to diagnose, and malformations range from subtle deformities, such as isolated fifth-finger clinodactyly, to complete absence of extremities, as seen in phocomelia syndromes such as Roberts-SC phocomelia and Al-Awadi Raas-Rothschild syndrome. Anomalies of the digits may be isolated or associated with other skeletal abnormalities. The differential diagnosis for abnormal fetal hands is vast and includes chromosomal abnormalities and genetic syndromes. Correctly identifying a hand anomaly is crucial because certain hand anomalies are strongly suggestive of specific disorders and can provide valuable clues to prenatal diagnosis.
One useful way of categorizing these is to separate the anomalies into six morphologic areas:
- 1.
alignment abnormalities (clenched hand, camptodactyly, clinodactyly, hypokinesia, clubhand, phocomelia)
- 2.
thumb anomalies
- 3.
abnormal size (macrodactyly, brachydactyly, trident configuration)
- 4.
abnormal echogenicity (under or overmineralized)
- 5.
abnormal number of digits (polydactyly, oligodactyly)
- 6.
constriction band sequence
- 1.
shape (clenched, clubhand, camptodactyly, clinodactyly, phocomelia, brachydactyly, trident hand)
- 2.
number of fingers (polydactyly, ectrodactyly, oligodactyly)
- 3.
thumbs (aplasia, hypoplasia, hitchhiker’s deformity, broad thumb, and triphalangeal thumb)
Disorder
Definition
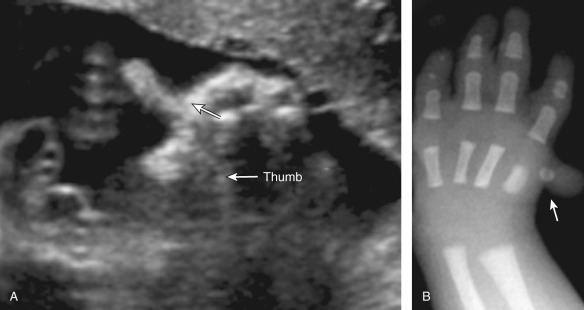
Hand abnormalities are associated with hundreds of different disorders. Briefly, abnormalities can affect the thumbs only and lead to hypoplasia, triphalangeal shape (three phalange bones instead of two), broadness, or abnormal angulation (hitchhiker’s thumb). Any of these abnormalities can occur in isolation, or they can be associated with a multitude of aneuploidies or genetic syndromes. Some abnormalities are nearly pathognomonic for certain disorders; for instance, a prenatal finding of hitchhiker’s thumb is strongly suggestive of diastrophic dysplasia ( Fig. 61.1 ). Broad thumbs are associated with the acrocephalosyndactylies (Apert, Carpenter, or Pfeiffer syndrome) and trisomy 13. Hypoplasia or aplasia of the thumb is seen in numerous disorders including Nager syndrome, radial aplasia-thrombocytopenia syndrome, Roberts-SC phocomelia syndrome, VATER ( v ertebral defects, imperforate a nus, t racheo e sophageal fistula, r adial and r enal dysplasia) association, and Townes-Brocks syndrome. Two diagnoses that should be strongly considered in the case of an absent or abnormal (hypoplastic or triphalangeal) thumb are Fanconi anemia and Holt-Oram syndrome.
Prevalence and Epidemiology
The frequency of the hand abnormalities, particularly the thumbs, is based on the incidence of the disorder. For example, the global prevalence of Fanconi anemia is estimated to be 1 : 1,000,000 to 5 : 1,000,000, affecting males and females and all races and ethnic groups. It is an autosomal recessive disorder, and the heterozygous mutation carrier frequency is estimated to be between 0.3% and 1%. There are at least 15 genes responsible for the different forms of Fanconi syndrome. The prevalence of Holt-Oram syndrome is estimated to be 1 : 100,000 total births. Holt-Oram syndrome is autosomal dominant, and approximately 85% of cases are attributed to new mutations in TBX5 .
Etiology, Pathophysiology, and Embryology
Fanconi anemia is a clinically and genetically heterogeneous disorder characterized by spontaneous chromosomal instability, visible during the DNA replication phase of the cell cycle as broken chromatids. These breakpoints can be misrepaired as chromatid interchanges, further contributing to chromosome damage. Fanconi anemia is also characterized by spontaneous arrest and delay during the G 2 stage of the cell cycle and by hypersensitivity to chromosome-breaking and antiproliferative effects of DNA cross-linking agents. The relationship between the defective genetic pathway of Fanconi anemia and the associated clinical features of the disease is not well understood at this time.
Limited data are available regarding the pathogenic processes that account for the manifestations of Holt-Oram syndrome. The TBX5 gene (member of the T-box family of transcription factor genes that is involved in forelimb and heart development) probably interacts with other transcription factors to produce the phenotype.
Manifestations of Disease
Clinical Presentation
Fanconi anemia is characterized by numerous physical and hematologic findings, although one-fourth of individuals have no physical abnormalities. In terms of prenatal diagnosis, besides abnormal thumbs, abnormalities of the cardiac, gastrointestinal, and renal systems can be associated with Fanconi anemia. Postnatal findings include cutaneous abnormalities such as café au lait spots, hyperpigmentation or hypopigmentation, short stature, microcephaly with a triangular face, small ear canals, and deafness. Hypogonadism and reduced fertility are associated with Fanconi anemia. Hematologic findings typically include pancytopenia with bone marrow failure in the first 10 years of life, with 90% experiencing bone marrow failure by the fourth decade of life. There is a significant risk of developing hematologic malignancies (predominantly acute myelogenous leukemia) and other nonhematologic malignancies of the head and neck, skin, gastrointestinal tract, and genital tract.
Holt-Oram syndrome is characterized by the presence of at least one upper limb preaxial radial ray malformation, including abnormal thumb (absent, hypoplastic, or triphalangeal) ( Fig. 61.2 ). It is autosomal dominant, with nearly 100% penetrance and variable expression of the phenotype. Family history of cardiac septation defects or atrioventricular conduction disease strengthens the suspicion of the diagnosis. Greater than 70% of individuals meeting the aforementioned strict diagnostic criteria have a mutation in the TBX5 gene, and the diagnosis can be confirmed through molecular genetic testing.










