, Marilyn J. Siegel2, Tomasz Miszalski-Jamka3, 4 and Robert Pelberg1
(1)
The Christ Hospital Heart and Vascular Center of Greater Cincinnati, The Lindner Center for Research and Education, Cincinnati, OH, USA
(2)
Mallinckrodt Institute of Radiology, Washington University School of Medicine, St. Louis, Missouri, USA
(3)
Department of Clinical Radiology and Imaging Diagnostics, 4th Military Hospital, Wrocław, Poland
(4)
Center for Diagnosis Prevention and Telemedicine, John Paul II Hospital, Kraków, Poland
Abstract
Tetralogy of Fallot (TOF) is characterized by four constant features: right ventricular outflow tract (RVOT) obstruction, malalignment (perimembranous) ventricular septal defect (VSD), biventricular origin of the aorta (overriding aorta), and right ventricular hypertrophy [1].
19.1 Tetralogy of Fallot
Tetralogy of Fallot (TOF) is characterized by four constant features: right ventricular outflow tract (RVOT) obstruction, malalignment (perimembranous) ventricular septal defect (VSD), biventricular origin of the aorta (overriding aorta), and right ventricular hypertrophy [1].
TOF is the most common cyanotic congenital heart defect, accounting for approximately 10 % of all congenital heart disease [2].
TOF is a conotruncal abnormality in which there is anterior deviation and underdevelopment of the outlet (infundibular) portion of the septum, resulting in narrowing and obstruction of the right ventricular outflow tract, an aortic root that overlies the right and left ventricles, and a large perimembranous VSD with the secondary development of right ventricular hypertrophy (Figs. 19.1 and 19.2) [3]. The RVOT obstruction is typically subvalvular (pulmonic infundibulum) stenosis, but it can be valvular or supravalvular and can occur at multiple levels (Figs. 19.1 and 19.3).
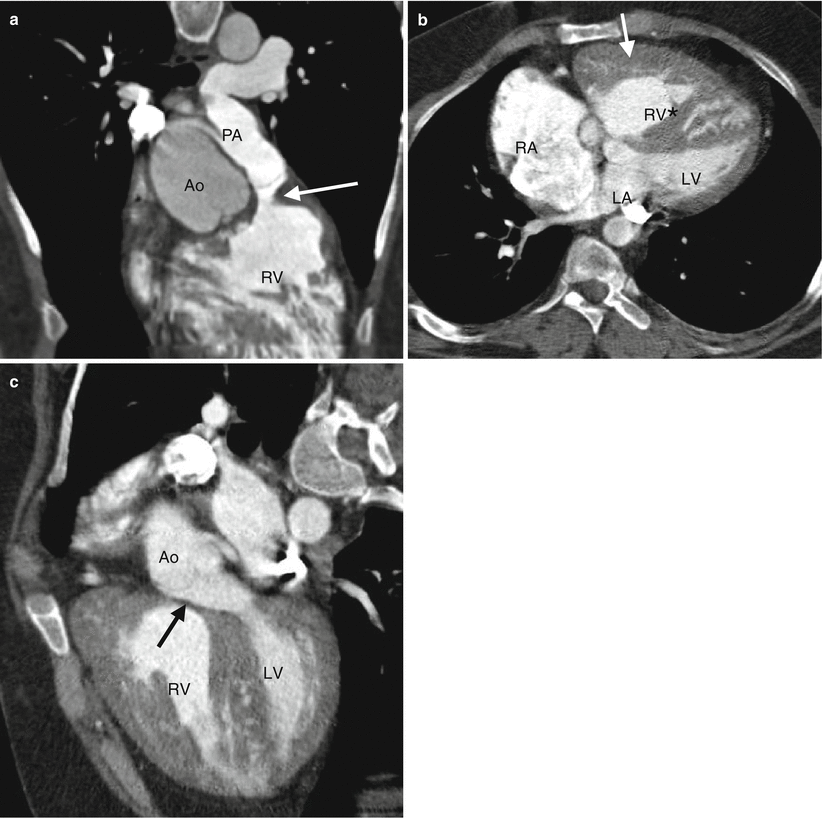
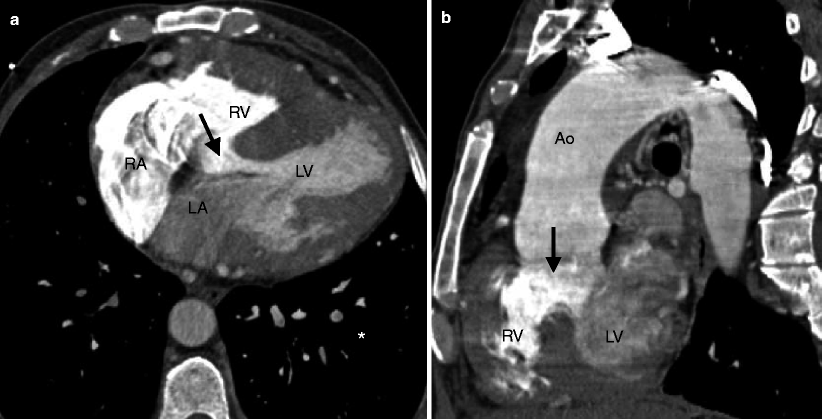
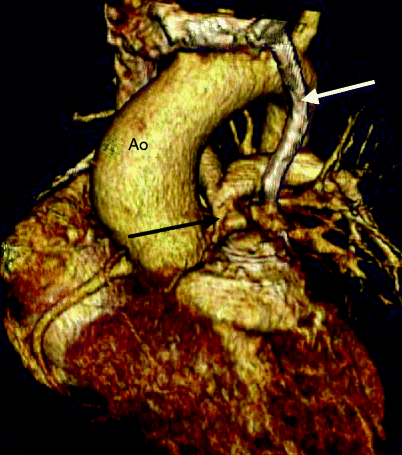
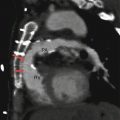
Fig. 19.1
Tetralogy of Fallot. A 34-year-old woman who underwent repair at age 6 years with a right ventricular outflow tract patch (patch across the pulmonary valve annulus) and VSD closure. Panel (a) is a coronal image showing residual subvalvular stenosis (arrow). Notice the dilated aortic root (Ao). Panel (b) is an axial scan showing right ventricular (RV) hypertrophy (arrow in panel b) secondary to the right ventricular outflow tract (RVOT) obstruction. Note the hypertrophied moderator band (asterisk). The VSD defect is closed. Panel (c) an oblique multiplanar reformatted image showing malalignment of the perimembranous septum (arrow), which has been repaired, and an overriding aorta (Ao). Again note the RV hypertrophy. LV left ventricle, PA pulmonary artery
Fig. 19.2
Tetralogy of Fallot. A 45-year-old man who has an unrepaired tetralogy of Fallot. Panel (a) is an axial image showing a large, nonrestrictive VSD (arrow) and right ventricular (RV) hypertrophy. Panel (b) is an oblique multiplanar reformat showing the malalignment of the unrepaired perimembranous VSD (arrow) and an overriding aorta (Ao). RV hypertrophy is again prominent. LV left ventricle, RA right atrium, LA left atrium
Fig. 19.3
Tetralogy of Fallot. A 32-year-old man who had a VSD closure and patch enlargement of the RVOT at age 5 years and returned with residual pulmonary valve stenosis. This image is a volume-rendered reformat and shows the severe narrowing at the level of the pulmonary valve (black arrow). White arrow: left superior vena cava
Infundibular pulmonic stenosis is mostly caused by overgrowth or hypertrophy of the septoparietal trabeculae. Hypoplasia of the pulmonary valve annulus, main pulmonary artery, and branch pulmonary arteries is common, and there may be branch pulmonary artery stenosis and peripheral pulmonary artery stenosis [1, 2]. Absence of the main pulmonary artery (left more often than right) may also coexist (Fig. 19.4). Other obstructions include a hypertrophied moderator band and apical trabeculation resulting in a two-chambered right ventricle [4] and infundibular or pulmonary valve atresia [5]. In the latter setting, the pulmonary arteries distal to the atresia are perfused via major aortopulmonary collateral vessels (MAPCAs) and/or the ductus arteriosus. In rare instances, the pulmonary arteries are supplied by a persistent fifth arch, an aortopulmonary window, or coronary artery fistulas [6–9]. Pulmonary atresia with VSD is discussed in Chap. 14; it is believed to result from right heart hypoplasia rather than maldevelopment of the outlet septum.
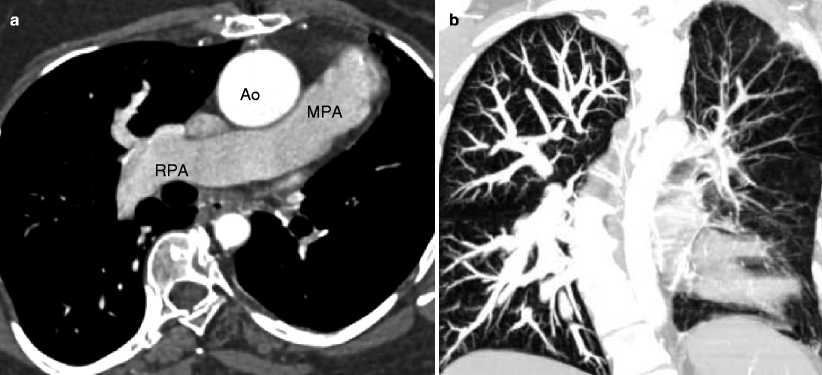
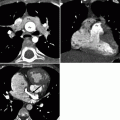
Fig. 19.4
Tetralogy of Fallot with absence of the pulmonary artery. A 32-year-old woman who had resection of infundibular muscle bundles, VSD closure, and augmentation of the right ventricular outflow tract and pulmonary artery with a transannular patch at age 7 years. Panel (a) is an axial scan showing the absence of the left pulmonary artery. The main (MPA) and right pulmonary arteries (RPA) are mildly dilated. Also note the dilated ascending aorta (Ao) caused by aortic insufficiency. Panel (b) is a coronal image using a lung window and shows that the left lung is smaller than the right lung and has less vascularity as well. Flow to the left lung was via multiple aortopulmonary collateral vessels
The overriding aorta, which lies above the ventricular septal defect, connects to both the right and the left ventricle. The aortic root can be displaced anteriorly or directly above the septal defect, but it is almost always abnormally located to the right of the pulmonary artery root. The aortic valve may originate predominantly from the left or right ventricle. The latter represents the coexistence of TOF with double-outlet right ventricle [3]. Aortic dilatation may occur due to aortic insufficiency (Fig. 19.5).
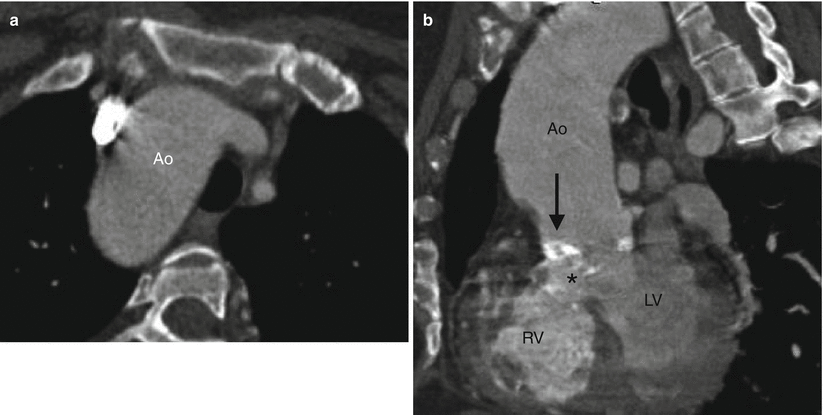
Fig. 19.5
Tetralogy of Fallot with right aortic arch and aortic root dilatation. A 44-year-old woman who underwent placement of a Waterston shunt at age 7 years and no additional surgical procedures. Panel (a) is an axial image showing the right aortic arch (Ao). The aorta is dilated due to aortic insufficiency. Panel (b) is a sagittal image showing dilatation of the aortic root (Ao) which overrides an unrepaired perimembranous VSD (asterisk). Also noted a calcified aortic valve (arrow), related to atherosclerotic disease
The malalignment VSD (also termed conal septal malalignment defect) is usually single, large, and nonrestrictive [3, 10]. Typically, it is located in the membranous septum in a subaortic position and is overridden by the aorta superiorly. Its superior margin is made up of the outlet septum, while its anterior margin is created by the fusion of the outlet septum with the anterior limb of septomarginal trabeculation. The posteroinferior part is formed by fibrous continuity between the leaflets of aortic, mitral, and tricuspid valves (80 % of cases) or by a muscular rim (20 % of cases) created by fusion of ventriculo-infundibular fold and the posterior limb of septomarginal trabeculation [11]. Rarely and most frequently in Asian and South American populations, the muscular outlet septum is completely absent and the interventricular communication represents a doubly committed, juxta-arterial VSD, which is also termed conal septal malalignment defect. The conal septal malalignment defect is also associated with conal septal hypertrophy [3, 12].
In 40 % of cases, TOF is associated with other associated abnormalities including right-sided aortic arch (Fig. 19.5), atrial septal defect (ASD), atrioventricular septal defect, anomalous coronary arteries (most frequently left anterior descending artery originating from right coronary artery) (Fig. 19.6), and persistent ductus arteriosus [1]. MAPCAs are also common and correlate with the degree of pulmonary stenosis or atresia.
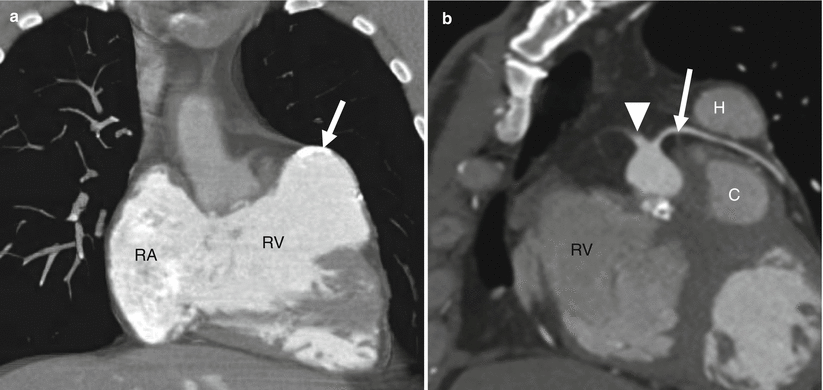
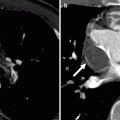
Fig. 19.6
Tetralogy of Fallot with a coronary artery anomaly. A 33-year-old man who had homograft repair of RVOT obstruction and a conduit placed from the homograft to the main pulmonary artery at age 2 years. Resection of the infundibular muscle was limited by a coronary artery coursing over the upper part of the right ventricular outflow tract just beneath the pulmonary valve annulus. Panel (a) is a depiction of the aneurysmal, partially calcified right ventricular outflow repair (white arrow). Panel (b) a sagittal oblique view demonstrating the right coronary artery (arrowhead) giving rise to the left anterior descending coronary artery (arrow), which courses between the homograft (H) and conduit (C) with no evidence of compression
The predominant clinical feature of most patients with TOF is cyanosis, and thus, most are diagnosed in infancy. Central cyanosis results from right-to-left shunting through a nonrestrictive VSD. The magnitude of shunting depends on the severity of obstruction at the RVOT, pulmonary valve, and/or pulmonary artery [10]. Surgical interventions are now performed in almost all neonates/infants with TOF, and the majority (>85 %) of adults have undergone primary repairs [13].
When the RVOT obstruction is mild, the degree of right-to-left shunting through the VSD will be less severe and physiology will be more similar to an isolated VSD. In this setting, patients may be acyanotic (so-called pink tetralogy) and the diagnosis will be made later in life. The clinical presentation in unrepaired adults may include cardiac murmur, right heart failure, or decreased exercise tolerance [2]. If the overriding aorta dilates, aortic regurgitation may develop and be a presenting symptom (Fig. 19.6).
Before advances in open heart surgery, palliative surgery in the form of a systemic-to-pulmonary artery shunt (Blalock–Taussig, Waterston, or Potts shunts) was performed prior to definitive repair (Chap. 22) [2]. With advances in surgical techniques, the current treatment is definitive repair in the first year of life. Most adults with TOF have undergone definitive repair, but occasionally patients reach adulthood having had only a palliative procedure. Definitive repair requires closure of the VSD and enlargement of the RVOT, relieving the outlet stenosis (Chap. 29) [2].
Postoperative complications (Chap. 29) of palliation include pulmonary artery distortion, pulmonary hypertension, left and right ventricular dysfunction, and congestive heart failure. Complications of definitive repair include aneurysmal dilatation of the RVOT and subsequent pulmonary regurgitation; stenosis at the anastomotic site of the pulmonary homograft, pulmonary valve, and/or main/branch pulmonary arteries; progressive aortic root dilatation and subsequent aortic regurgitation; biventricular failure; and residual VSD.
19.1.1 Cardiac Computed Tomography (CT) in the Assessment of Tetralogy of Fallot
In patients with unoperated TOF, CT can demonstrate the following morphologic findings needed to plan patient management: (a) the ventricular (right and left ) morphology including the wall thickness, size, and systolic function (contraction, volume); (b) the morphology and size of both atria; (c) the anatomy, size, and presence/magnitude of obstruction(s) of the RVOT, pulmonary valve, main pulmonary artery, and branch arteries; (d) morphology of the overriding aorta (size and degree); (e) the size and location of VSD(s); (f) the anatomy of the cardiac valves; (g) aortic arch sidedness; and (h) the presence of coexisting anomalies including ASD, patent ductus arteriosus, aortopulmonary collaterals, and coronary artery anomalies [14–17]. Of high importance is the presence of a coronary artery branches crossing the RVOT, which can have an impact on the surgical strategy (Fig. 19.6). In addition, the presence of coexisting atherosclerotic coronary artery disease should be evaluated. See Table 19.1.
Table 19.1
CT assessment in unrepaired tetralogy of Fallot
Morphology, wall thickness, size, and systolic dysfunction of both ventricles |
Morphology and size of atria |
Anatomy, size, and presence/magnitude of obstruction(s) of RVOT, pulmonary valve, main pulmonary artery, and branches |
Anatomy and size of ascending aorta, including assessment of aortic root dilatation |
Presence, size, and morphology of VSD |
Anatomy of the cardiac valves |
Side of the aortic arch |
Coexisting anomalies, including atrial septal defect, patent ductus arteriosus, aortopulmonary collaterals, and coronary artery anomalies |
Of note, cardiac CT provides only limited information regarding valvular function. The assessment of pulmonary valve stenosis/regurgitation and aortic and tricuspid valve regurgitation is especially important and requires performance of echocardiography and cardiac magnetic resonance imaging.
The postsurgical assessment of these patients is discussed in Chap. 29.
19.2 Double-Outlet Ventricles
When both great arteries arise from one ventricular chamber, the ventriculoarterial connection is termed “double outlet.” In double-outlet ventricles, the aorta and pulmonary artery may arise from the right ventricle (double-outlet right ventricle) or from the left ventricle (double-outlet left ventricle). Rarely, both arterial trunks arise from both ventricles. This arrangement is termed double-outlet both ventricles.
19.2.1 Double-Outlet Right Ventricle
Double-outlet right ventricle (DORV) is a heterogeneous group of congenital heart defects in which both the aorta and main pulmonary artery arise entirely or predominately from the morphologic right ventricle [18]. DORV is present when more than 50 % of the both arterial trunks arise from the morphologic right ventricle.
DORV accounts for 1–5 % of all congenital heart diseases with incidence of 1 per 10,000 births [19].
The basic morphology of DORV is malposition of the great arteries with a ventricular septal defect (VSD) (Fig. 19.7). There is great variability in the anatomy of the DORV. DORV represents the part of arterial override continua, when
[3, 20, 21]
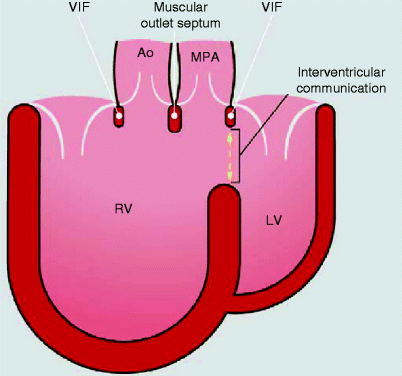
(a)
Overriding aorta transits from tetralogy of Fallot (ToF) or simple ventricular septal defect with overriding aorta to DORV configuration
(b)
Overriding pulmonary artery transits from discordant ventriculo-arterial connection with ventricular septal defect to DORV configuration with subpulmonary interventricular communication
(c)
Overriding aorta and pulmonary artery transit from double outflow left ventricle to DORV configuration
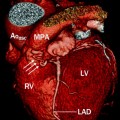
Fig. 19.7
Diagram shows both the aorta (Ao) and main pulmonary artery (MPA) arising from the right ventricle (RV). LV left ventricle, VIF ventriculo-infundibular fold
The pathophysiologic classification of DORV is based on defining the position of the VSD relative to the arrangement of the great arteries. The VSD can be subaortic, subpulmonic, doubly committed (immediately beneath the semilunar valves), or noncommitted (remote from both semilunar valves) (Fig. 19.8). Most commonly, the VSD is single and nonrestrictive, but restrictive and multiple connections may occur [18]. Pulmonary and systemic blood flow and saturations are determined by the ratio of pulmonary to systemic resistance, which is related to the relative positions of the VSD and great arteries, the presence of pulmonary stenosis, and the amount of right ventricular mixing.
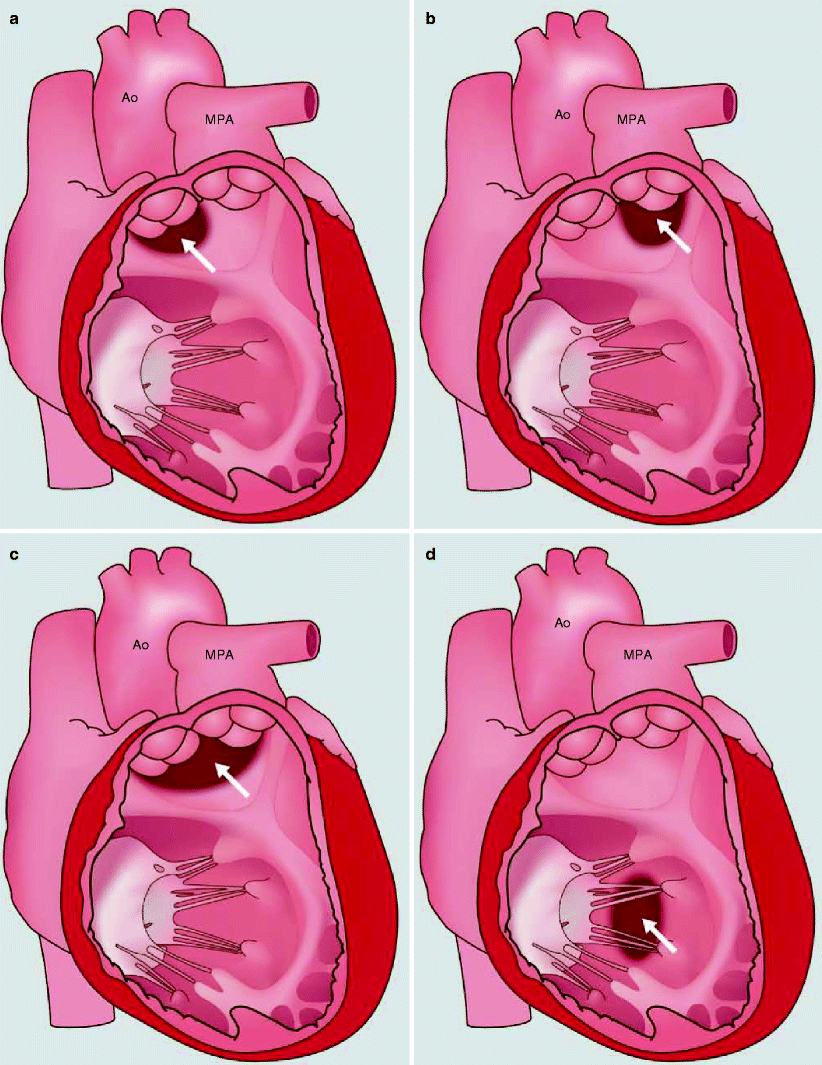
Fig. 19.8
The variants of interventricular communication in double-outlet right ventricle. See text for a detailed description. (a) subaortic, (b) subpulmonary, (c) doubly committed, and (d) noncommitted (remote). Ao aorta, MPA main pulmonary artery. See text for description. White arrows point to the ventricular septal defect
Subaortic VSD-type (tetralogy of Fallot-type) DORV accounts for more than 50 % of DORV cases (Fig. 19.9). The great arteries are normally related with the aortic origin posterior and to the right of the pulmonary origin [22, 23]. Because the normal spatial relationship is maintained, left ventricular outflow is directed toward the aorta, resulting in aortic oxygen saturations that are greater than those of the pulmonary artery. Pulmonary stenosis is present in up to 50 % of patients. In these patients, the physiology resembles that of tetralogy of Fallot, where the aorta completely overrides the right ventricle. In the absence of pulmonary stenosis, the physiology resembles that of a large isolated VSD [20]. This anatomy may result in congestive heart failure. Subaortic VSD is associated with L-transposition of the great arteries and an anomalous course of the right coronary artery, which crosses the pulmonary outflow tract [18].
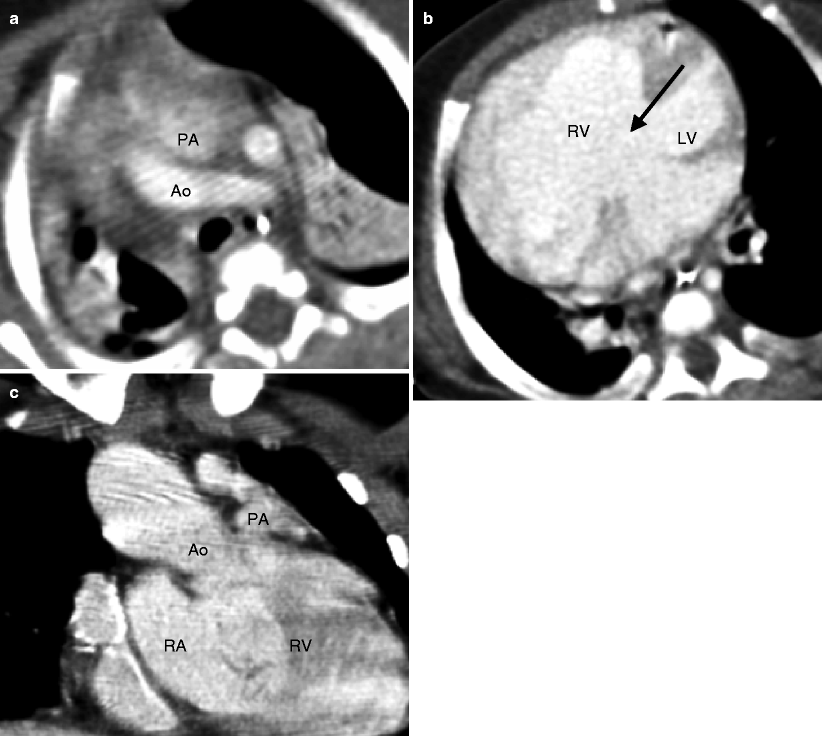
Fig. 19.9
Subaortic VSD-type double-outlet right ventricle. The great arteries are normally related. Panel (a) is an axial scan showing the aorta posterior and to the right of the pulmonary artery. Panel (b) is an image showing the large ventricular septal defect (arrow). Panel (c) is a coronal view showing the aorta and the pulmonary artery arising from the right ventricle (RV). Most of the ventricular outflow is directed toward the aorta, which is larger than the pulmonary artery. Ao aorta, PA pulmonary artery, LV left ventricle, RV right ventricle
Subpulmonary VSD-type DORV (so-called Taussig–Bing anomaly) is encountered in 30 % of patients with DORV [22–25]. The left ventricular outflow is directed toward the pulmonary artery, resulting in pulmonary artery saturations greater than aortic saturations. The great artery relationship is transposed [18]. The aortic and pulmonary origins have either a parallel arrangement (positioned side by side) or the aorta is to the right and slightly anterior to the pulmonary artery (Figs. 19.10 and 19.11). In the absence of pulmonary stenosis, the physiology is similar to that of transposition of the great arteries. If there is associated pulmonary stenosis, the physiology is similar to that of tetralogy of Fallot. The subpulmonary form of DORV is associated with subaortic stenosis and aortic arch obstruction (aortic coarctation and interrupted aortic arch). It is also associated with straddling and cleft mitral valves [18, 22, 23, 26, 27].
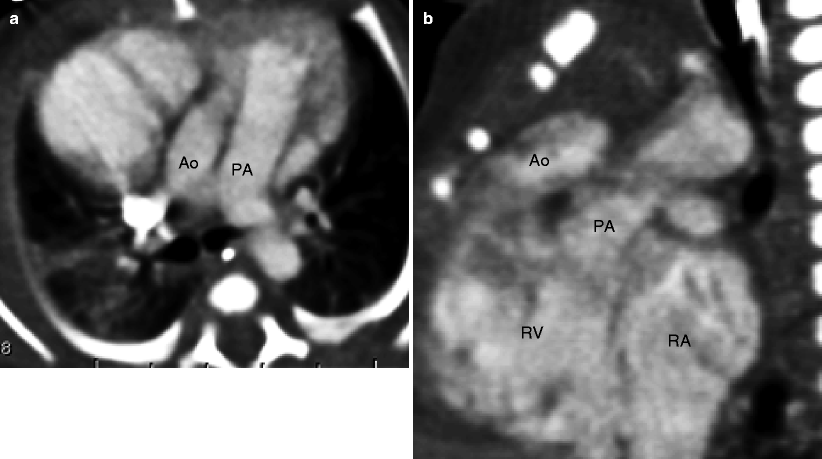
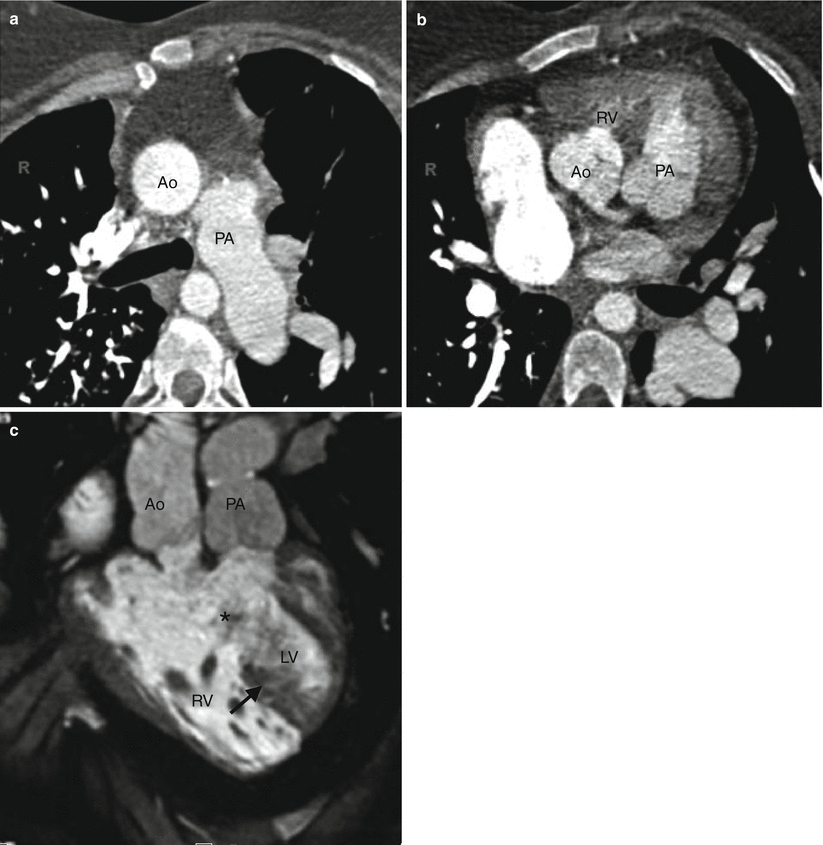
Fig. 19.10
Subpulmonic VSD-type double-outlet right ventricle. Panel (a) is an axial scan showing the aorta and pulmonary artery in a parallel arrangement (positioned side by side). Panel (b) is a sagittal scan showing the aorta and the pulmonary artery arising from the right ventricle (RV). Most of the ventricular outflow is directed toward the pulmonary artery. Ao aorta, PA pulmonary artery, RV right ventricle, RA right atrium
Fig. 19.11
Subpulmonic VSD-type double-outlet right ventricle in a 31-year-old woman with a prior Fontan operation. Panel (a) is an axial view showing the aorta anterior and to the right of the pulmonary artery (D-transposition). Panel (b) is an axial scan and panel (c) is a coronal oblique cut. Both images demonstrate the two great arteries arising from the hypertrophied and dilated right ventricle (RV), medial and to the right of the interventricular septum (arrow in panel c), consistent with double-outlet right ventricle. Panel (c) demonstrates the very large VSD (asterisk)
Doubly committed VSD-type DORV occurs in 10 % of cases of DORV [25]. The left ventricular outflow is equally directed to the aorta and pulmonary artery. The great arteries are normally related [18]. Since the left ventricular outflow is equally shared by the aorta and pulmonary artery, the pathophysiology resembles that of a VSD.
Noncommitted or remote VSD-type DORV occurs in 10 % of DORV cases. The anatomy and physiology is similar to that of an isolated VSD or atrioventricular canal defect. Most commonly, the great arteries are normally related [24, 25].
Rarely, the interventricular connection will be absent, and DORV is then accompanied by a small atrial septal defect and hypoplasia of the mitral valve and left ventricle [28]. Due to commitment of both arteries to the right ventricle outlet (i.e., infundibular), the septum becomes a right ventricular structure.
Associated anomalies include situs abnormalities (dextrocardia, situs inversus, atrial isomerism), atrioventricular valve abnormalities (mitral stenosis, cleft mitral valve, straddling mitral and/or tricuspid valve, common atrioventricular valve), ventriculoarterial connection and outflow tract abnormalities (subaortic stenosis, atrioventricular discordance, atrioventricular valve atresia), atrial septal defects, and single ventricle [29–31]. Extracardiac anomalies include anomalous venous return, aortic coarctation, interrupted aortic arch, and persistent ductus arteriosus [18]. Coronary artery abnormalities are also common, including anomalous origin of the right coronary artery (RCA) from the left main coronary artery (LMCA), duplication of left anterior descending coronary artery (LAD), anomalous origin of LAD or left circumflex artery from the RCA, and single right or left coronary artery. The presence of pulmonary and aortic outflow tract obstructions can influence the pathophysiology of the heart malformation.
The clinical findings vary with the anatomy and include cyanosis, heart failure, and pulmonary hypertension. Most patients with DORV are diagnosed in the first month of life and undergo palliative repair (pulmonary artery banding or Blalock–Taussig shunt) or surgical repair. The surgical interventions depend on the location of the VSD, the size of the left ventricle, the type of ventriculoarterial connection, and the type of pulmonary blood flow (restricted on unrestricted) [18].
Definitive repairs vary and include intraventricular repair using a tunnel (baffle) to direct left ventricular flow to the aorta, arterial switch operation to connect the left ventricle to the neoaorta in combination with VSD closure, and univentricular-type repair, using a bidirectional Glenn shunt (superior vena cava to pulmonary artery) [32–36]. Rarely, unoperated patients survive to adulthood. The subaortic and doubly committed VSDs are often suitable for an intraventricular repair. The subpulmonic or “Taussig–Bing” anomaly with physiology similar to transposition may be treated with an arterial switch operation. The noncommitted VSD may require a Fontan procedure.
Potential complications after surgical repair include residual or recurrent VSD, residual or recurrent outflow tract obstructions, and atrioventricular valve regurgitation. Complications of univentricular repair include stenosis/narrowing of the Glenn shunt and Fontan procedure.
19.2.2 Double-Outlet Left Ventricle
Double-outlet left ventricle (DOLV) is a congenital anomaly in which both the aorta and main pulmonary artery arise entirely or predominately from the morphologic left ventricle [37].
DOLV is a very rare anomaly. It accounts for approximately 5 % of patients with double-outlet ventricle and its incidence is estimated to be less than 1 in 200,000 live births [38].
Similar to DORV, DOLV is characterized by heterogeneous morphology and clinical presentations. The VSD’s location may be subaortic, subpulmonary, doubly committed, or noncommitted (remote) [37, 39–41]. Subaortic and subpulmonic conal figurations are most common. DOLV occurs most often with atrial situs solitus and with atrioventricular concordance.
Subaortic VSD-type DOLV is present in 75 % of all DOLV patients. Pulmonary outflow tract obstruction is common, occurring in 90 % of cases. The great arteries are usually abnormally related and the aorta is usually anterior to the pulmonary artery [39, 41]. A right anterior aorta is more frequent than a left anterior aorta. DOLV with subaortic VSD with normally related great arteries is very rare [39]. The clinical presentation can resemble that of tetralogy of Fallot or complete transposition depending on the degree of pulmonary stenosis.
Subpulmonary VSD-type DOLV occurs in 15 % of cases [41]. Aortic outflow tract obstruction (aortic hypoplasia, aortic coarctation, or interrupted aortic arch) is common, occurring in about 80 % of cases [41]. The clinical manifestation is that of pulmonary overcirculation, similar to that of a large VSD.
Doubly committed VSD-type DOLV is present in 10 % of cases [41]. The VSD is overridden by both the main pulmonary artery and aorta. Of note, due to the varying degree of aortic and pulmonary artery override, the distinction between DOLV and DORV may be problematic, and thus, this anomaly has been referred to as double-outlet both ventricles [42]. In most cases, the great arterial relationship is side by side. Aortic or pulmonic outflow tract obstruction is rare.
Noncommitted (remote) VSD-type DOLV is extremely rare [43]. Pacifico et al. described one patient with DOLV with a posterior inlet VSD and a left-sided, anterior aorta.
Most patients with DOLV present in the neonatal period with cyanosis due to pulmonary outflow obstruction or with heart failure due to aortic outflow obstruction or a large VSD. Palliation or surgical repair is done early in life. Rare unoperated patients survive to adulthood.
Surgical repairs include a left ventricle-to-pulmonary outflow tract closure, rerouting of blood from the right ventricle to pulmonary artery using patch reconstruction, pulmonary root or pulmonary artery translocation onto the right ventricle with or without Lecompte maneuver, Rastelli-type procedure, or Fontan procedure and VSD closure [37]. In general, DOLV with two relatively well-developed ventricles is often repaired by left ventricle-to-pulmonary outflow tract closure, a Rastelli conduit from the right ventricle to pulmonary artery, or pulmonary root translocation and VSD closure. DOLV with a functionally single-ventricle or with tricuspid valve atresia usually requires a Fontan procedure.
19.2.3 Cardiac Computed Tomography (CT) in the Assessment of Both DORV and DOLV
CT allows a comprehensive heart evaluation including (a) the morphology, size, geometry, and systolic function of both ventricles; (b) location of the interventricular connections and relationships to the atrioventricular junction; (c) morphology and anatomic relationships of the ventriculoarterial connections, including the conal configuration as well as the degree of great vessel override/commitment to the ventricles; (d) presence, morphology, size, and extent of obstruction of the pulmonary and aortic outflow tracts; (e) the spatial relationship of the great arteries; (f) atrial size and morphology; (g) the anatomy of the cardiac valves; (h) evaluation of aortic arch anatomy; and (i) the presence of coronary artery and other coexisting abnormalities. See Table 19.2.
Table 19.2
Cardiac CT assessment in double-outlet right and left ventricles
Morphology, size, wall thickness, geometry, and systolic function of both ventricles |
Morphology and anatomic relation of atrioventricular junction connections to VSD |
Morphology and anatomic relation of ventriculoarterial junction (conotruncal abnormalities and arterial override/commitment to ventricles) |
Presence, anatomy, size, and magnitude of aortic and/or pulmonary outflow tract obstructions |
Anatomy, size, and spatial relationship of aorta and pulmonary artery |
Morphology and size of atria |
Anatomy and function of cardiac valves |
Evaluation of coronary artery and aortic arch anatomy |
Other anomalies |
Post-interventional imaging assessment should include evaluation of (a) ventricular morphology, size, and systolic function; (b) the anatomy, size, and presence/magnitude of residual aortic and pulmonary outflow tract obstruction(s) and extracardiac conduits (obstruction/leak); (c) the presence of residual interventricular communication; (d) the anatomy, size, course, and spatial arrangement of the aorta and pulmonary artery, with attention to aortic root dilatation and residual obstructions in the aortic arch; (e) the anatomy of the atrioventricular valves; (f) atrial morphology and size; (g) coronary artery anatomy with attention to compression/stenosis; and (h) coexisting anomalies [14, 15]. See Table 19.3.
Table 19.3
CT assessment of post-interventional complications in double-outlet ventricles
Left and right ventricular dilatation and dysfunction |