Some brain tumors results are interesting due to their rarity at presentation and overwhelming imaging characteristics, posing a diagnostic challenge in the eyes of any experienced neuroradiologist. This article focuses on the most important features regarding epidemiology, location, clinical presentation, histopathology, and imaging findings of cases considered “bizarre.” A review of the most recent literature dealing with these unusual tumors and pseudotumors is presented, highlighting key points related to the diagnosis, treatments, outcomes, and differential diagnosis.
Key points
- •
The most likely diagnosis of a hemorrhagic pineal lesion associated with elevated serum β-HCG levels in CSF is pineal choriocarcinoma (PCCC).
- •
Meningioangiomatosis (MA) is a rare epileptogenic pseudotumor that in a majority of the cases shows calcifications and cortical invasion mimicking a malignant meningioma.
- •
Diffuse midline glioma (DMG) comprises 80% of all brain stem tumors in children and 80% of them are malignant.
- •
Papillary glioneuronal tumor (PGNT) is a rare cystic mass with an enhancing mural nodule located in the temporal lobes.
- •
Diffuse leptomeningeal glioneuronal tumor (DLGNT) is a very rare and aggressive disease with fatal outcome.
Introduction
This article discusses the most important features regarding epidemiology, prevailing location, clinical presentation, histopathology, and imaging findings of cases that the authors consider interesting due to their rarity. New nomenclature related to these tumors is included, according to the 2016 World Health Organization (WHO) central nervous system (CNS) tumor classification. A review of the most recent literature dealing with these unusual tumors and pseudotumors is presented, highlighting key points related to the diagnosis, treatments, outcomes, and differential diagnosis.
Introduction
This article discusses the most important features regarding epidemiology, prevailing location, clinical presentation, histopathology, and imaging findings of cases that the authors consider interesting due to their rarity. New nomenclature related to these tumors is included, according to the 2016 World Health Organization (WHO) central nervous system (CNS) tumor classification. A review of the most recent literature dealing with these unusual tumors and pseudotumors is presented, highlighting key points related to the diagnosis, treatments, outcomes, and differential diagnosis.
Pineal choriocarcinoma
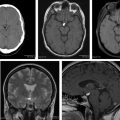
PCCC is a rare malignant nongerminomatous germ cell tumor (GCT) and most aggressive form of the gestational trophoblastic disease that has an exceedingly low survival rate when compared with other GCTs. PCCC is always mixed with other elements of GCTs and because they contain syncytiotrophoblasts (β-HCG–producing cells) they result in high levels of HCG in blood and CSF. Due to their location, they cause hydrocephalus despite a small size. A key imaging feature of PCCCs is the presence of intratumoral hemorrhage, which correlates with a poor prognosis.
Epidemiology
Most patients with primary intracranial PCCC are young men (3–22 years of age) who experience precocious puberty. Mean age of presentation is 11.8 years with a male-to-female ratio of 74:19.
Location
Choriocarcinoma arising within or outside the uterus after pregnancy is referred to as gestational choriocarcinoma. Nongestational choriocarcinoma arises from germ cells in gonadal or extragonadal locations (mediastinum, retroperitoneum, and pineal gland), generally in the midline.
Clinical Presentation
In the majority of patients, the clinical presentation depends on the tumor location. There are no specific symptoms related to this tumor. However, headache, vomiting, nausea, visual impairment, polydipsia, polyuria, and endocrinologic abnormalities are among the most common reported symptoms.
Imaging Findings
Most nongerminomatous GCTs reported are case reports with nonspecific imaging findings. Intracranial choriocarcinomas and specifically PCCC, however, tend to appear on CT as ovoid, heterogeneous, slightly hyperdense, and relatively well-defined lesions with lobulated margins centered in the pineal gland region ( Fig. 1 ). At the time of diagnosis, tumor size ranges from 2.0 cm to 4.5 cm. MRI offers better characterization of the tumor than CT. Tumors and their metastases are highly vascular and intratumoral hemorrhage is typical. On T1-weighted images, they demonstrate high signal due to presence of subacute blood. As a result, multiple areas of heterogeneous hypointensity and hyperintensity are seen on T2-weighted images. Susceptibility-weighted imaging (SWI) demonstrates blooming artifact within the tumor due to presence of hemoglobin products.
Histopathology
PCCCs are characterized by presence of stromal vascular channels that form blood lakes and hemorrhagic necrosis. Recent publications suggest that the connections between tumor-formed sinusoids and blood vessels might be a cause of hemorrhage in PCCC.
Treatment
PCCC is highly resistant to standard treatments. The first option is total tumor resection even in absence of hydrocephalus. In the recent years, many investigators have used a combination of total tumor removal, chemotherapy, and radiation therapy with satisfactory results. Despite these therapeutic strategies, the disease is usually fatal.
Differential Diagnosis
Because hemorrhage is a key feature of PCCC and hemorrhagic lesions centered in the pineal region are exceptional, the differential diagnosis can be narrowed to a few entities, including hemorrhagic metastasis, vascular malformation, cavernous malformation, and hemorrhagic pineal cyst. In addition, other tumors in the pineal region with intratumoral hemorrhage include pineocytoma, meningioma, hemangiopericytoma, embryonal carcinoma, and ganglioglioma. Nevertheless, the presence of elevated β-HCG level in serum and/or CSF favors the diagnosis of choriocarcinoma.
Prognosis
When the diagnosis is confirmed before the lesion develops hemorrhage, gross total resection has been demonstrated to improve hydrocephalus and prognosis. In the presence of hemorrhage, rebleeding rate is high as is the risk of massive hemorrhage and, for this reason, prompt and total tumor resection followed by radiotherapy and chemotherapy is recommended to prevent fatal outcomes.
Extraventricular neurocytoma
CNs are rare benign tumors usually located in the lateral ventricles near the foramina of Monro but can also occur in the third and fourth ventricles. The name CN reflects its midline location within the ventricular system, but when these tumors arise in the brain parenchyma they are called EVNs. EVN shares similar biological behavior and histopathologic characteristics with CN. These neurocytomas were included in the 2000 WHO classification of tumors of the CNS but it was not until 2007 when they were classified as separate entities. EVNs have a more aggressive biological behavior and a poorer outcome than CNs. CNs as well as EVNs may mimic several entities, such as oligodendrogliomas, ependymomas, and astrocytomas. For this reason, they can pose a diagnostic challenge. Immunohistochemistry, however, has greatly improved diagnostic accuracy.
Epidemiology
EVN occur with almost equal sex distribution with a slight female predominance. Their exact incidence is unclear, with fewer than 100 cases reported in the literature to date. EVN typically affects children and young adults during the third to fourth decades. Mean age of presentation is approximately 27 years.
Location
EVNs have been reported in most of the nervous system but they tend to arise in the periventricular regions. In adults, EVNs occur in the frontal lobes, followed by temporal, parietal, and occipital lobes. In children, the spine is a common location after the frontal and temporal lobes.
Clinical Presentation
Most common symptoms include increased intracranial pressure, seizures, gait disturbances, vision changes, headaches, and vomiting. Symptom onset varies from 3 months to 10 years.
Imaging Findings
EVNs do not have specific imaging characteristics and appear as solid well-circumscribed lesions involving the deep white matter or the cortical gray matter of the cerebral hemispheres. EVNs demonstrate a wide range of appearances, including perilesional edema and heterogeneous contrast enhancement ( Fig. 2 ). They contain areas of calcification, cystic degeneration, and hemorrhage. Even though nonenhancing EVNs resembling low-grade gliomas have been described, the former generally are hypointense or slightly hyperintense on T1-weighted images. Magnetic resonance spectroscopy (MRS) shows a nonspecific pattern with prominent choline and decreased or absent NAA. Perfusion is usually increased in the solid components of the tumor, which may indicate atypical features.
Histopathology
Histologic features of EVNs are small round neoplastic cells with round, regular nuclei embedded in a matrix of fine neuronal processes. Unlike the uniform morphologic pattern of CNs, EVNs show sheetlike patterns, clusters, ribbon-like appearance, or Homer-Wright rosettes. EVNs differ from CNs in their pronounced tendency for astrocytic, typically pilocytic features and/or ganglionic differentiation, which can be seen in 50% to 60% of cases. It may be difficult to differentiate EVN from oligodendroglioma, but, unlike the latter, EVNs are strongly immunoreactive for synaptophysin, both in the neuropil and in perinuclear cytoplasm. EVNs that have an MIB-1 index greater than 2%, focal necrosis, increased vascularity, and mitoses are called “atypical neurocytomas,” resulting in higher rates of mortality and recurrences than typical EVNs.
Treatment
Extent of resection is the key determinant of recurrence and gross total excision is the best prognostic indicator leading to better local control and survival rates, but total excision is often limited by proximity to eloquent areas. Radiotherapy has demonstrated good results in local control but no improvement of overall survival.
Differential Diagnosis
Oligodendrogliomas comprise the most challenging differential diagnosis due to their similar appearances and histopathologic features. Immunohistochemistry has improved the differentiation of oligodendrogliomas from EVNs. Although a specific genetic profile has not been found in EVNs, some investigators have described the presence of codeletion of 1p19q in approximately 25% of EVNs with atypical features and infiltrative growth patterns. Recent studies have demonstrated that EVNs lack p53 overexpression, α-internexin positivity, O6-methylguanine DNA methyltransferase (MGMT) promotor methylation, and isocitrate dehydrogenase (IDH)1/IDH2 mutation.
Other differential diagnoses include pilocytic astrocytoma, PXA, and ganglioglioma. A diagnosis of EVNs can be aided by imaging features that demonstrate well-demarcated borders rather than infiltrating edges.
Prognosis
EVNs are benign neoplasms in nature, except for atypical neurocytomas, which display an aggressive behavior and have overall a poor prognosis with a high rate of recurrence. According to some reports, extreme age, especially younger age, is associated with atypical features and higher risk for recurrence with a worse outcome.
Pleomorphic xanthoastrocytoma with intraventricular extension
PXA is an uncommon, generally benign astrocytic neoplasm first described as a variant of cerebral astrocytomas that demonstrates extensive involvement of the leptomeninges. PXA is a WHO grade II neoplasm. However, 9% to 20% of PXAs undergo malignant transformation when they recur after resection and some show anaplastic features at diagnosis. Moreover, several investigators suggest the existence of high-grade PXA or at least the existence of a PXA with aggressive presentation. Recently, the 2016 WHO classification included a different type of PXA with aggressive behavior, numerous recurrences, shorter survival times, and poor outcome compared with the more benign variant. Anaplastic PXA (aPXA) WHO grade III is the nomenclature given to this entity, as an alternative to PXA with anaplastic features. To date, more than 20 cases that fulfill the criteria for aPXA have been reported. Primary meningeal aPXAs and leptomeningeal dissemination of intraparenchymal aPXA have also been described. A parenchymal mass with intraventricular extension is an exceedingly rare presentation of PXA, with only a few case reports in the English literature. The following discussion focuses on the classic presentation of PXA.
Epidemiology
PXA generally arises in children and young adults with an incidence rate of 0.07:100,000. PXAs can arise at any decade of life with a mean age of 26 years. Most data about these tumors come from case reports and small series, reflecting their rarity. Currently, there are not enough cases to determine gender or racial predilection.
Location
The classic presentation is that of mass with a dominant intra-axial component, localized superficially in the cerebral hemispheres. Approximately 98% of cases reported are supratentorial. The most common location is the temporal lobes, followed by the parietal, frontal, and occipital lobes. It is typically superficial, consistently involving the leptomeninges, infiltrating the underlying parenchyma, and extending into the perivascular spaces. Less common locations include the cerebellum, spinal cord, retina, and sellar region.
Clinical Presentation
The most common presentation is chronic epilepsy. There are few reports of PXAs with CSF dissemination at the time of diagnosis.
Imaging Findings
Although PXA is associated with a wide range of imaging appearances due to its inherent “pleomorphic” histopathology, presence of a cystic tumor with an enhancing mural nodule and adjacent inner skull table scalloping are common findings. Perilesional edema and intratumoral hemorrhage have also been reported ( Fig. 3 ). Scarce data on diffusion-weighted MRI suggest that relatively low apparent diffusion coefficient (ADC) values are found in PXA compared with other peripheral low-grade supratentorial neoplasms.
Differential Diagnosis
PXA should be included in the differential diagnosis of cortical-based cystic lesions with a solid enhancing mural nodule or neoplasms with extensive involvement of the leptomeninges in young patients. Differential includes ganglioglioma, pilocytic astrocytoma, glioblastoma (GB), oligodendroglioma, and dysembrioblastic neuroepithelial tumor (DNET). Most DNETs and oligodendrogliomas do not enhance. Occasionally, different entities may mimic a cortical-based enhancing mass and could be considered in the differential (eg, meningioma, sarcoid, or lymphoproliferative mass).
Histopathology
Hallmarks of PXAs include pleomorphism with dense reticulin network and lipid deposits in neoplastic cells. Almost every PXA shows glial fibrillary acid protein (GFAP)-positive cells and S100 immunoreactivity. aPXA shows malignant histologic features with aggressive clinical behavior. A PXA can be considered anaplastic if 5 or more mitoses per 10 high-power fields and necrosis are present.
Treatment
Treatment of choice is gross total resection, which is the most important predictor of recurrence but not of overall survival. Although PXA has a benign behavior, there is a tendency to recur if incompletely excised. Because of the rarity of the tumor, the role of adjuvant radiotherapy or chemotherapy remains uncertain.
Prognosis
Despite their highly pleomorphic cytology, PXA has a relatively favorable prognosis. Nevertheless, advanced patient age and CSF dissemination at presentation are poor prognostic factors. A significant decrease in overall survival is seen with aPXA compared with PXA.
Meningioangiomatosis
MA is a rare meningovascular malformation of the CNS first described as an incidental autopsy finding in a patient with neurofibromatosis (NF) type 2. It is characterized by presence of an epileptogenic plaque-like or nodular mass in the leptomeningeal and cortex. Approximately 25% to 50% of MAs are associated with NF type 2 whereas the remaining cases occur sporadically. Multiple lesions have also been reported. Several publications report MA in association with oligodendroglioma, cerebral hemorrhage, arteriovenous malformations, and meningeal hemangiopericytoma, with meningiomas the most commonly associated neoplasms. It does not become malignant or recur. Its pathogenesis remains unclear. Consequently, MA is usually misdiagnosed and occasionally mistreated by unsuccessful methods.
Epidemiology
MA occurs in children and young adults with no gender predilection.
Location
Most MAs are supratentorial and involve the cerebral cortex (90%), particularly in the frontal or temporal lobes. Other locations, including third ventricle, thalamus, cerebral peduncles, and brainstem, have been reported. Some publications suggest that when MA is associated with NF type 2 there is a frontal lobe tendency.
Clinical Presentation
Patients with meningioangiomatosis typically present with seizure disorders, which are the exclusive clinical problem in the majority of cases. Seizures are usually simple or complex partial, and tend to be refractory to treatment. Persistent headache is the second most common symptom and usually disappears after surgical resection of the lesion.
Imaging Findings
Imaging findings vary according to the histopathologic components. Gliosis and edema can be seen in the periphery of the lesion on CT and MRI. Regardless of location, MA is always well defined intra-axially or extra-axially but occasionally may have ill-defined margins as a result of cortical invasion. The utility of CT is to detect calcifications, which have been reported in up to 89.6% of cases. On MRI, the lesion is classically isointense to hypointense on T1-weighted imaging (T1WI) and hypointense to hyperintense on T2-weighted imaging (T2WI). A most striking characteristic of MA is presence of a gyriform hyperintensity on fluid-attenuated inversion recovery (FLAIR), which corresponds to thickened cortex with proliferating leptomeningeal vessels interwoven with bands of fibrous connective tissue. SWI identifies calcifications missed on CT. Occasionally, MA can be divided into 2 imaging patterns: solid and a cystic appearances (the solid variant being more common). On T1 postcontrast sequences, various patterns of enhancement have been reported, including avid, mild, or no enhancement. Most of cases, however, demonstrate prominent homogenous contrast enhancement ( Fig. 4 ).
Histopathology
The hallmark of MA is leptomeningeal calcification and meningovascular proliferation intermingled with fine bands of fibrous connective tissue. MA involves the cerebral cortex and subcortical white matter.
MA has a low MIB-1 index. Neurofibrillary tangles are not unusual. Its pathogenesis is controversial but a hamartoma with degenerative changes, leptomeningeal meningioma with invasion in adjacent brain tissue, and cortical vascular malformation have been proposed. Kim and colleagues found that the meningiomatosis portions of meningiomatosis-meningioma have loss of heterozygosity at the 22q12 locus in 28.6% of coexisting cases of meningiomatosis-meningioma, whereas each pure meningiomatosis harbors 1 loss of heterozygosity at either 22q12 or 9p21. Approximately 90% of cases show cortical invasion, but findings like atypia, mitoses, and necrosis are not found. Immunohistochemical staining of MA shows markers, such as epithelial membrane antigen, S-100 protein, and GFAP.
Treatment
Treatment of choice is total surgical removal, which is important for seizure control and pathologic diagnosis. An accurate diagnosis is important, especially when MA is associated with an underlying meningioma because a misinterpretation of atypical or malignant meningioma may result in incorrect treatment.
Differential Diagnosis
Differential diagnosis includes oligodendroglioma, ganglioglioma, dysembryoplastic neuroepithelial tumor, low-grade astrocytoma, meningioma, granulomatous meningitis, and parasitic diseases. The main differential diagnosis of MA-associated meningioma to be considered is a malignant meningioma with an invasion of the brain.
Prognosis
Excellent results are obtained after total surgical removal because most patients became free of seizure and recurrence after surgical treatments. Antiepileptic drug administration, however, is required in greater than 70% of patients.
Intraventricular glioblastoma
GB is the most common and most malignant primary brain tumor of adults. Despite the different locations and patterns, IVGB is extremely rare. GB is classified as WHO grade IV due to the presence of hypercellularity, nuclear polymorphism, brisk mitotic activity, prominent microvascular proliferation, and/or necrosis. IVGB is thought to arise from the neuroglial cells of the septum pellucidum or fornix or may be secondary to an abnormal healing process in the subependymal zone.
Epidemiology
In the United States, GB accounts for 15.1% of all primary brain tumors and for 46.1% of primary malignant brain tumors. It is more common in older adults (65–84 years), 1.6 times more common in men than in women, and has prevalence 2 times higher among whites compared with blacks. IVGBs are found in younger patients (19–47 years).
Location
IVGBs are rare, with only 30 cases reported to date. Most IVGBs are located in the frontal horns or ventricular bodies ( Fig. 5 B, C). The third ventricle is an extremely rare location.
Clinical Presentation
Symptoms are vague, including headaches, urinary incontinence, gait disturbances, blurred vision, vomiting, and confusion. Symptoms do not develop until the tumor reaches a size large enough to cause obstructive hydrocephalus or compression of surrounding structures.
Imaging Findings
Imaging features of IVGB are similar to those located in extraventricular locations. On CT they present as mixed density masses with contrast enhancement of the non-necrotic areas. On MRI, T1WIs shows an infiltrative mass with irregular borders and mixed signal intensity. T2 sequences show surrounding edema. T1 postcontrast images show heterogeneous or ringlike contrast enhancement surrounding the necrosis. On perfusion images, high rCBV could be seen within the solid portions of the tumor (see Fig. 5 ). One case of IVGB with minimal enhancement has been reported.
Differential Diagnosis
Differential diagnosis includes ependymoma, subependymoma, subependymal giant cell astrocytoma, and choroid glioma.
Histopathology
GBs are classified into IDH wild-type (90%) or IDH mutant (10%) and non-otherwise-specified (NOS) GBs in which IDH evaluation cannot be performed. Presence of extensive necrosis and endothelial cell proliferation raises the category to WHO grade IV. Microscopic features show pleomorphic fibrillary astrocytes, gemistocytes, and bipolar bland-appearing but mitotically active small cells. High proliferation index (MIB-1) exceeding 10% is typical. IVGB shares all these features.
Treatment
Treatment includes surgical resection followed by radiation with a total dose of at least 54 Gy and thereafter chemotherapy. Transcallosal and transcortical are the surgical approaches most commonly used.
Prognosis
Survival for GB is low, with only 5% of patients surviving 5 years postdiagnosis. IVGBs have a slightly better prognosis than parenchymal GBs due to their presentation in younger patients, with a median survival of 25 to 35 weeks after surgery.
Embryonal tumor with multilayered rosettes
ETMRs are thought to originate from primitive or undifferentiated brain cells. Embryonal tumors other than medulloblastoma had undergone some changes in their terminology in the 2016 CNS WHO classification. The term, primitive neuroectodermal tumor (PNET) , has been removed and they are classified according to the presence of amplification of the C19MC region on chromosome 19 (19q 13.42). The presence of C19 MC amplification results in a diagnosis of ETMRs (ETMR-C19MC-altered). In its absence, the tumor is called ETMR-NOS.
Epidemiology
The incidence of ETMRs peaks between 0 and 4 years. They account for less than 1% of brain tumors in children. ETMRs are rare, with an incidence less than 5% of all supratentorial tumors in children. Only 20% to 30% of these tumors occur in adults, with fewer than 100 ETMRs reported in the literature among this group. In adults the age of presentation is between the second and third decades.
Location
Cerebral hemispheres are the most common location with equal distributions among the frontal, temporal, and parietal lobes. Other locations, such as suprasellar region, have been reported. At the time of presentation, the tumors usually measure more than 5 cm.
Clinical Presentation
Symptoms are related to the size and location, including weakness, headache, vomiting, seizures, and changes in personality.
Imaging Findings
ETMRs are usually large masses, more than 5 cm in size at the time of presentation ( Fig. 6 A). They are well-delineated masses with absent or minimal peritumoral edema despite their large size. Typically they are heterogeneous isodense to hyperdense masses on CT. Calcifications are seen in 70% of cases. On MRI, the signal on T1/T2 is variable due to presence of blood products, calcifications, and cystic changes. FLAIR shows areas of necrosis (hyperintense) and cystic components (hypointense) ( Fig. 6 ). On diffusion-weighted imaging, the tumor shows high signal and reduced ADC, due to high cellular density. The solid components demonstrate avid heterogeneous contrast enhancement. Perfusion MRI shows increased relative cerebral blood volume. ETMRs develop subarachnoid dissemination in 40% of cases; thus, the entire neuroaxis should be imaged (see Fig. 6 G, H). The presence of metastases changes management, requiring chemotherapy and increased radiation dose.