- •
Thickened, doming aortic valve.
- •
Ascending aorta may be dilated in isolated aortic stenosis; however, aortic hypoplasia is common when aortic stenosis is seen in the presence of associated left ventricular anomalies.
- •
Measure aortic annulus diameter.
- •
Assess flow pattern across aortic valve looking for turbulence of flow and peak velocity in order to determine gradient.
- •
Assess for aortic insufficiency.
- •
Look for associated defects such as subaortic stenosis, mitral valve abnormalities.
- •
Assess left ventricular endocardium for “echo-brightness” suggesting endocardial fibroelastosis.
- •
Assess left ventricular function.
- •
Aortic arch size and presence or absence of coarctation of the aorta.
- •
Shunting across the atrial septum (if right to left, there is normal left atrial and left ventricular compliance; if left to right, left atrial pressure and left ventricular compliance may be abnormal and may predict poor left-sided function and/or viability).
Anatomy and Anatomical Associations
Aortic stenosis (AS) is a disease in which egress from the left ventricle (LV) into the aorta is obstructed by an abnormality along the left ventricular outflow tract (LVOT). The obstruction can occur at any of three levels: the subvalvar region, across the aortic valve itself, or in the supravalvar region. The severity of AS is related to the degree of obstruction across the LVOT.
Discrete stenosis at the level of the aortic valve is the most common cause of LVOT obstruction. Abnormal flow across a bicuspid aortic valve, a condition in which two of the coronary cusps fuse together, is a frequent finding in AS. Other causes of valvar AS include abnormalities of the commissures between the aortic cusps, abnormalities of the leaflets themselves, or abnormalities of the valve annulus ( Figure 19-1 ).
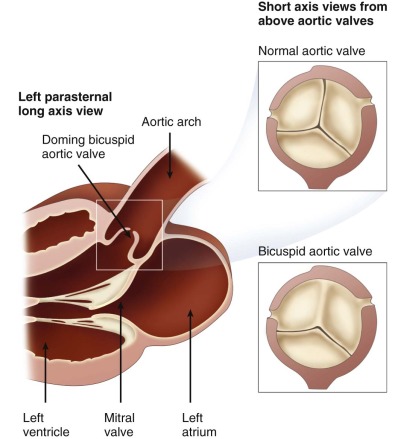
In the case of a bicuspid aortic valve, fusion may be complete or incomplete and the degree of obstruction may be inconsequential or may be so severe as to be of hemodynamic significance. The aortic valve is composed of leaflets or cusps, and commissures, or joining points/borders between the leaflets. There are three cusps, or leaflets, present in the aortic valve, designated based on their relationship to the origin of the coronary arteries—the right cusp (most anterior), the left cusp, and the noncoronary cusp (most posterior). Fusion of the right and left coronary cusps accounts for approximately 85% of bicuspid aortic valves. Fusion of the right and noncoronary cusps is the next most common, whereas fusion of the left and noncoronary cusps is rare.
Single cups, unicuspid, or unicommissural aortic valves, although far less common than bicuspid aortic valves, are much more likely to present with significant obstruction early in life. These valves have only a slitlike opening along the unaffected commissure, causing a significant reduction in the effective cross-sectional orifice of the outflow tract. Another mechanism of obstruction is partial fusion of all three commissures. In these valves, there are no discrete aortic leaflets and the degree of obstruction is related to the size of the small central orifice as the leaflets attempt to separate and open during systole. Myxomatous aortic valves are unusual but may also be responsible for AS because the dysplastic nature of the leaflets prevents full excursion. Hypoplasia of the aortic annulus is another potential cause of obstruction based on the inherent smallness of the outflow tract. This either can occur with structurally normal valve leaflets or can be seen in association with valvar abnormality such as commissural fusion, as well.
Valvar AS is often found in isolation, although 20% of cases may have associated cardiac defects. These commonly include ventricular septal defect, a patent ductus arteriosus, or coarctation of the aorta. Progressive aortic root dilation is a common finding in patients with bicuspid aortic valves.
Subvalvar AS is responsible for 8% to 20% of cases of congenital LVOT in infants and children. It is often associated with the combination of a posteriorly malaligned infundibular septum and a ventricular septal defect. It may also be associated with mitral stenosis or left-sided obstructive lesions including coarctation of the aorta and interrupted aortic arch. Patients with complete common atrioventricular canals may have subaortic stenosis as a result of atrioventricular valve attachments obstructing LVOT.
Secondary subaortic stenosis may complicate other forms of congenital heart disease. Patients with ventricular septal defects may develop subvalvar AS as a result of the development of a subaortic membrane. These are extremely rare in the fetus and newborn, and typically appear at approximately 2 to 3 years of age. Likewise, patients with hypertrophic cardiomyopathy may develop subvalvar AS as a result of asymmetrical septal hypertrophy.
Supravalvar AS is the least common type of AS and is most often associated with Williams’ syndrome, which accounts for up to 50% of all cases. Non-Williams syndrome defects in the elastin gene account for the vast majority of the remainder of supravalvar AS cases.
Frequency, Genetics, and Development
Bicuspid aortic valve is the most common congenital cardiac abnormality found in the human species, with a prevalence of approximately 1.3%. Most bicuspid aortic valves do not cause obstruction in childhood or adolescence, but the incidence of AS increases dramatically through adulthood as the bicuspid leaflets thicken and calcify. It is estimated that AS as a whole accounts for approximately 4% of all congenital heart disease in the pediatric population. For isolated AS, the male-to-female ratio is reported to be in the range of 3 : 1.
Development of the aortic valve occurs early in gestation. The process begins after the aortopulmonary septum divides the bulbus cordis and the truncus arteriosus into two distinct vessels, the aorta and the pulmonary artery, at around 6 weeks of gestation. The aortic valve leaflets form from outpouchings of three subendocardial ridges near the orifice of the aortic valve. The subendocardial ridges go through a process of remodeling until they form the thin, pliable leaflets of the aortic valve. The three aortic cusps are attached to a fibrous ring at the valve annulus and, in normal development, are equal to each other in size. Alterations in the development of the aortic valve leaflets may result in valvar forms of AS.
The genetic basis for AS is likely multifactorial and is not completely delineated. Turner’s syndrome (46 X,O) has been associated with AS as well as other forms of left-sided obstruction including coarctation of the aorta and hypoplastic left heart syndrome (HLHS). The 11q terminal deletion disorder has also been associated with AS. Other nonsyndromic forms of AS may be passed on in a familial manner, sometimes as a single, autosomal dominant mutation, or may be sporadic. When isolated AS occurs as part of a familial cluster, other left-sided obstructive lesions are often found in affected relatives. Mutations in NOTCH-1 have been implicated as a specific genetic anomaly responsible for some cases of aortic valve malformations.
Prenatal Physiology
In the fetus, isolated AS is well tolerated. Even in the case of HLHS, in which the LV does not develop into a normal-sized chamber, fetal growth and development is relatively unaffected. In the normal fetal circulation, the LV is responsible for pumping the small amount of pulmonary venous blood that returns from the lungs as well as the highly oxygenated blood that streams from the ductus venosus across the foramen ovale. These two blood pools represent 45% of total cardiac output and have the highest concentration of oxygen. The LV pumps this blood into the ascending aorta to the head and neck vessels, thereby supplying the brain and the coronary arteries with the most highly oxygenated blood in the fetus. In cases of AS, blood flow redistributes away from the LV such that it is responsible for a smaller amount of the overall cardiac output. Depending on the degree of stenosis, this may result in partial perfusion of the head and neck vessels in a retrograde manner from the right ventricle through the ductus arteriosus and into the aortic arch. This does not cause fetal distress, but the impact of mild alteration in the delivery of oxygen to the brain during fetal development is unknown.
Prenatal Management
Depending upon the severity of AS, there may be an alteration in left ventricular compliance as the ventricle hypertrophies. There will then be less right-to-left shunting at the atria level, which will diminish left ventricular filling.
Upon echocardiographic evaluation, the direction of shunting at the atrial level should be assessed. In cases of isolated AS, but in which the LV is deemed to be adequate in size and makeup to function as the systemic ventricle, there will be the normal right-to-left direction of shunting, or bidirectional shunting. Evidence for left-to-right atrial level shunting suggests the possibility of an inadequate LV and present, or evolving, HLHS.
Echocardiography should assess the nature and velocity of flow across the aortic valve. In AS, the flow will be turbulent, nonlaminar flow. The peak systolic velocity will exceed 1.5 m/sec, but may not reach the very high levels seen after birth, because flow across the LV is limited and further distributed away from the LV in the presence of an abnormal aortic valve. Even in severe cases of AS, typical values for peak velocity reach 3 m/sec, but not much higher.
Investigation of the ascending aorta for the appearance of root dilation can help in the diagnosis of mild cases of bicuspid aortic valve. A complete assessment of the aortic arch for transverse arch hypoplasia and coarctation of the aorta is essential.
In prenatal life, the right ventricle is able to compensate for any amount of diminished output from the LV without affecting fetal growth and development. However, although AS does not affect global fetal growth, there is the potential for the arrest of fetal growth of the LV. In these cases, some centers have undertaken a strategy of intrauterine balloon dilation of the aortic valve to try to encourage left ventricular ejection to promote left ventricular growth (see discussion of hypoplastic left heart syndrome, earlier).
Postnatal Physiology
After the closure of the ductus arteriosus, usually within the first 72 hours of life, the right ventricular contribution to systemic cardiac output is eliminated. The LV takes on the sole responsibility of pumping oxygenated blood returning from the lungs across the aortic valve and out to the body. In cases of mild or moderate AS, the obstruction may be well tolerated through the neonatal period and into childhood. However, in cases of severe or critical AS ( critical is defined as requiring ductal support for systemic perfusion), neonatal intervention is often necessary to relieve the obstruction. Without early intervention, infants with severe or critical AS may present in shock and may not survive the neonatal period. Catheter-based balloon aortic valvuloplasty allows neonates to survive the neonatal period without surgery and may allow for the growth of an undersized LV. Newborn infants with critical AS may, interestingly, not have a significantly high gradient across the aortic valve as blood is shunted away from the LV, and systemic perfusion occurs via the patent ductus arteriosus, shunting right to left (pulmonary artery to aorta) in systole.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


