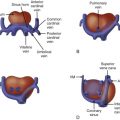
- •
Investigate for heart size (cardiothoracic ratio).
- •
Investigate systolic function, qualitative and quantitative (myocardial performance index, shortening fraction).
- •
Look for evidence of pericardial effusion or hydrops.
- •
Look for atrioventricular valve regurgitation.
- •
Assess Doppler signals of ventricular inflow, ductus venosus, and umbilical vein.
- •
Measure peak systolic middle cerebral artery velocity when looking for fetal anemia as a possible explanation for cardiomegaly and high-output state.
- •
Calculation of combined cardiac output and serial follow-through can be instructive in cases of markedly low or markedly elevated cardiac output values.
- •
Assess Doppler flow patterns from umbilical artery and middle cerebral artery. Ratio of pulsatility indices reflects flow and overall fetal wellness or unwellness.
Fetal cardiomyopathy is a disorder in which the heart muscle of the fetus is diseased and not functioning properly. It makes up approximately 2% to 4% of all forms of cardiovascular disease seen in the fetus. Fetal cardiomyopathy can be classified into two categories. Dilated cardiomyopathy (DCM) is defined as the presence of ventricular chambers enlarged above normal and poor systolic function. Hypertrophic cardiomyopathy (HCM) is defined as abnormally thickened ventricular walls, in the absence of any structural abnormality that might explain the hypertrophy. Cardiomyopathy in the fetus can be due to a variety of causes such as infection, metabolic disease, genetic disorders of myocardial dysfunction, fetal anemia, or it is of unclear etiology and, therefore, labeled as idiopathic. Some examples of diseases causing fetal cardiomyopathy are listed in Table 35-1 ; however, a complete, exhaustive list is not possible because the potential etiologies and pathways to myocardial dysfunction are endless.
| CATEGORY | CAUSE |
|---|---|
| Dilated Cardiomyopathy | |
| Infection |
|
| Metabolic/genetic |
|
| Anemia |
|
| Other |
|
| Hypertrophic Cardiomyopathy | |
| Metabolic/genetic |
|
| Other |
|
Dilated Cardiomyopathy
Etiologies
Infectious etiologies are the most likely cause for DCM in the fetus, with metabolic/genetic abnormalities a close second. Viral agents can cross the placenta and cause active fetal viral myocarditis. The presence of an isolated large pericardial effusion in association with a dilated, poorly functioning heart indicates ongoing myocarditis and inflammation. Maternal history for an acute illness may be elicited, but commonly, no such event can be documented upon questioning. In viral myocarditis, dysfunction may resolve spontaneously or persist, suggesting myocardial damage.
Mutations in the tafazzin gene (TAZ) located on the X chromosome are responsible for a variety of X-linked cardiomyopathies that may present in the fetus. Barth’s syndrome is one such disorder in which there is DCM, cyclical neutropenia, and growth delay. Isolated noncompaction of the left ventricle (NCLV) can occur in an X-linked manner or may be seen without any particular genetic/chromosomal explanation. In this disorder, there are deep crypts within the left ventricle (LV) apex and free wall, with the noncompacted region measuring at least twice the diameter of the compacted myocardium. It is a disease primarily of the LV because the right ventricle (RV) is normally noncompacted. NCLV manifests a wide range of clinical symptoms with some patients asymptomatic with good ventricular function and others with poor systolic function. NCLV can evolve over time into DCM with progressive wall thinning, ventricular dilation, and dysfunction.
Fetal anemias, either through viral suppression as with parvovirus or genetic hemoglobinopathy, can cause DCM. Hemoglobin Bart’s disease is the homozygous form of alpha-thalassemia in which oxygen-carrying capacity is severely diminished. Hemoglobin Bart’s develops in fetuses with four-gene deletion alpha-thalassemia, in which no alpha chain of hemoglobin is produced. The gamma chains produced during fetal development combine to form gamma chain tetramers, which transport oxygen poorly. Most individuals with four-gene deletion thalassemia and consequent hemoglobin Bart’s manifest severe hydrops fetalis and die in utero. This disorder was characterized at St. Bartholomew’s Hospital in London; hence, the name hemoglobin Bart’s. When hemoglobin Bart’s disease is present, severe fetal hydrops and demise are nearly certain and can cause a “mirror syndrome” in the mother, placing her at risk as well.
A rare, but striking, disorder known as infantile arterial calcinosis can be identified during fetal life. Fetal imaging demonstrates the appearance of a thick, echo-bright “icing” of calcification outlining the walls of the great vessels. The disease is characterized by extensive calcification of medium and large arteries. Histologically, calcifications are seen to extend from the internal elastic lamina into the intima and media and are associated with giant cell reaction and smooth muscle proliferation. The aorta, main pulmonary arteries, or renal arteries can be affected. Myocardial dysfunction is common, presumably related to coronary calcification and myocardial ischemia. In addition, hypertension due to renal artery disease compounds the pathophysiology. Most cases lead to fetal hydrops and demise, or some fetuses make it to term with multiorgan system involvement but then manifest neonatal demise. Rarely, milder forms can be identified in infancy and may respond to chelating agents and diphosphonates.
Although most known for its effects on the conduction system, maternal autoimmune disorders such as lupus or Sjögren’s disease can cause DCM. Inflammation of the myocardium with dilation and dysfunction can occur in conjunction with destruction of conduction tissue or independently. Testing for SS-A and SS-B antibodies is warranted as part of the workup for DCM because most mothers carrying such fetuses can be completely asymptomatic without any hint of autoimmune disease. We, as well as others, have noticed an interesting echocardiographic phenomenon that seems to be indicative of possible maternal autoimmune disease. Some fetuses exposed to these antibodies will develop a unique echo-brightness to various regions of the myocardium, either at the atria, at the atrioventricular groove, or at the crux of the heart. We suspect that this may represent ongoing regional inflammation, which leads to an increased ultrasound reflectivity.
Prenatal Diagnosis
In general, the fetus with DCM will exhibit dilation of the left, right, or both ventricles, with poor systolic function. Qualitative cardiomegaly can be seen upon fetal echocardiography and the cardiothoracic ratio will exceed 40%. Shortening fraction measurement can provide a quantitative sense of the degree of systolic dysfunction and will be less than 30%. Atrioventricular valve regurgitation is common, due to either annular dilation and poor valve leaflet coaptation or papillary muscle dysfunction. Pericardial effusion, in particular with infectious causes, can be seen. Diastolic abnormalities may occur simultaneously or, at times, precede the onset of systolic dysfunction. Abnormalities such as fusion of the diastolic inflow pattern across the tricuspid or mitral valves, absence or reversal of flow with atrial contraction in the ductus venosus, or venous pulsations all reflect elevated ventricular filling pressures. Global measures of myocardial function are helpful in quantifying the degree of dysfunction. The myocardial performance index will be elevated, typically greater than 0.5, and can be used serially to assess for progression of disease. We have used the Doppler-derived combined cardiac output as a means to evaluate for the overall impact of dysfunction. Normal combined cardiac output is approximately 400 to 500 mL/kg/min of flow. In our experience, we have found that a value below 400 mL/kg/min reflects a decompensated state and suggests severe disease with poor outcome. Hydrops fetalis when associated with DCM indicates markedly elevated ventricular filling pressures and low cardiac output. The presence of hydrops fetalis bodes poorly for outcome, with a high likelihood of fetal or neonatal demise. The detection of any ventricular dilation and dysfunction requires close serial evaluation because disease progression can be rapid in some cases.
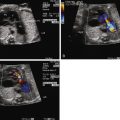
Evaluation of the arterial vascular systems, the umbilical arterial (UA) and middle cerebral arterial (MCA) flow, offers important information in the fetus with DCM. As an indirect measure of the adequacy of cardiac output, the resistance of the MCA should normally always be higher than the UA, which reflects a healthy distribution of flow. Hence, MCA pulsatility index (PI) should be higher than UA PI. When cardiac output is diminished, there is a vasoregulatory attempt to increase blood flow to the cerebral circulation, and the MCA PI will drop below the UA PI value. Serial evaluation of the UA and MCA PI values is, therefore, an important component of the overall fetal echocardiographic assessment of DCM. Furthermore, in conditions of fetal anemia, the MCA peak systolic velocity has been reported to be markedly elevated and can be a reliable measure of the degree of anemia ( Figure 35-1 ). As oxygen-carrying capacity diminishes with reduction in hemoglobin, there is a compensatory increase in cerebral volume of flow reflected by an increase in MCA peak systolic velocity. Assessment of MCA peak systolic velocity is of significant value because it can also be incorporated into the differential diagnostic assessment when trying to determine the etiology of DCM.

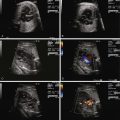
A useful scoring system for grading severity of fetal congestive heart failure has been developed. The cardiovascular profile score (CPS) comprises five categories of findings that, if abnormal, portray the degree of heart failure ( Figure 35-2 ). A normal heart would have a complete score of 10, and points are subtracted based on findings present. The categories include measures of degree of hydrops, venous Doppler, heart size, heart function, and umbilical arterial Doppler tracings. Studies have demonstrated the utility of the CPS in predicting outcome and correlation with other measures of cardiac performance such as the Tei index. In our view, the CPS can be helpful, but we would caution the use of a general “heart failure” score when applying it for a specific disease process because each disease has its own unique pathophysiology. For example, in twin-twin transfusion syndrome, the pathophysiology and severity of disease may not lend itself toward findings that are specifically present in the CPS. Findings such as wall thickness and right-sided outflow tract obstruction, key elements of twin-twin transfusion syndrome, are not included in the CPS. In general, all of the elements of the CPS can be helpful tools to gauge the degree of fetal unwellness. However, it should not be used exclusively in the absence of other variables that may provide greater information concerning the state of a specific disease.