, Marilyn J. Siegel2, Tomasz Miszalski-Jamka3, 4 and Robert Pelberg1
(1)
The Christ Hospital Heart and Vascular Center of Greater Cincinnati, The Lindner Center for Research and Education, Cincinnati, OH, USA
(2)
Mallinckrodt Institute of Radiology, Washington University School of Medicine, St. Louis, Missouri, USA
(3)
Department of Clinical Radiology and Imaging Diagnostics, 4th Military Hospital, Wrocław, Poland
(4)
Center for Diagnosis Prevention and Telemedicine, John Paul II Hospital, Kraków, Poland
Abstract
Patent ductus arteriosus (PDA) is the persistent communication between the descending aorta and main or proximal left pulmonary artery [1].
15.1 Patent Ductus Arteriosus
15.1.1 Definition
Patent ductus arteriosus (PDA) is the persistent communication between the descending aorta and main or proximal left pulmonary artery [1].
15.1.2 Epidemiology
Isolated PDA accounts for 5–10 % of congenital heart diseases with an incidence of 0.5 in 1,000 live term births [2].
15.1.3 Morphology
The patent ductus arteriosus is a vascular structure that connects the proximal descending aorta to the main or left pulmonary artery just distal to the left subclavian artery (Figs. 15.1, 15.2, and 15.3). During fetal life, the ductus arteriosus is a normal structure that allows most of the blood leaving the right ventricle to bypass the pulmonary circulation and enter the descending aorta effectively bypassing the developing lungs. Functional complete closure usually occurs within 24–48 h of birth in term neonates, although permanent closure does not occur until 2–3 weeks of life, and is related to fibrous band proliferation of the intimal lining of the duct. The resulting fibrous persists as the ligamentum arteriosum. Persistence of ductal patency in term infants after the neonatal period is considered abnormal [3]. Of note, failure of ductus arteriosus contraction in preterm neonates is not uncommon and is thought to be due to poor prostaglandin metabolism that stems from lung immaturity.
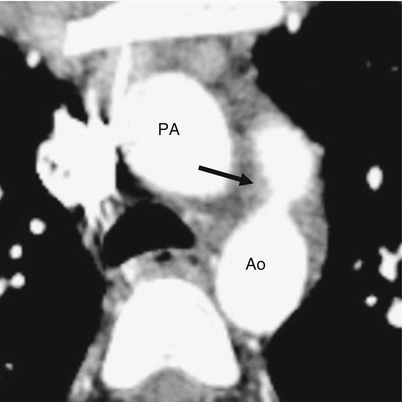
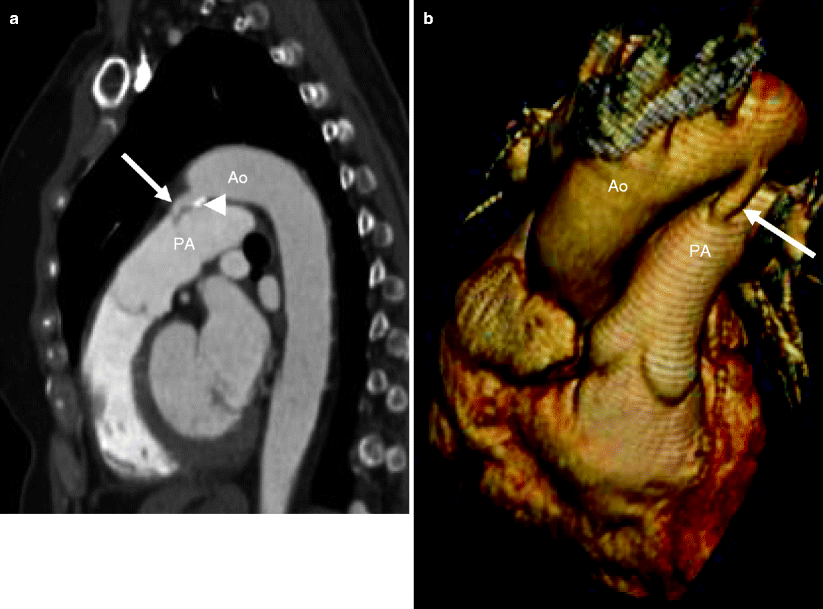
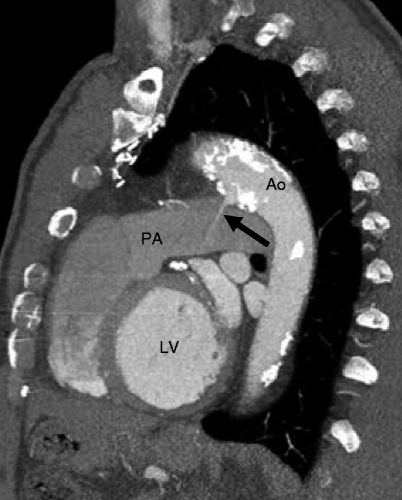
Fig. 15.1
Patent ductus arteriosus. An axial contrast-enhanced computed tomogram illustrating a patent communication (arrow) between the pulmonary artery (PA) and proximal descending aorta (Ao)
Fig. 15.2
A 65-year-old female with a known patent ductus arteriosus presenting with chest pain. Panel (a) is a sagittal view and panel (b) is a 3D oblique coronal reconstruction. Each shows a communication (arrow) between the aorta (Ao) and main pulmonary artery (PA). The duct has a wide open aortic ampulla (13 mm diameter) and narrows at its junction with the pulmonary artery (3 mm diameter) (type A, Krichenko classification). Also note calcification at the ductal-aortic junction (arrowhead in panel a)
Fig. 15.3
Patent ductus arteriosus. A 65-year-old man with history of a repaired patent ductus arteriosus as a teenager. Here, a sagittal reconstruction shows a jet of contrast-enhanced blood (arrow) flowing from the aorta (Ao) to the pulmonary artery (PA). The PDA is large and short (type B, Krichenko classification) LV left ventricle
Most typically, a PDA is left-sided but it can be right-sided or bilateral. Although a left ductus arteriosus is a normal structure during normal fetal development, the presence of a right ductus arteriosus is usually associated with other congenital heart abnormalities most typically involving the aortic arch or conotruncal area.
15.1.4 Coexisting Abnormalities
In adults, a PDA is usually an isolated finding. Rarely, it is associated with other cardiac abnormalities. The most frequent association is an atrial septal defect or ventricular septal defect [1, 4]. Other associated cardiovascular anomalies include aortic coarctation, interrupted aortic arch, hypoplastic left heart and pulmonary atresia with ventricular septal defect (ductus-dependent lesions), tetralogy of Fallot, and discordant ventriculoarterial connections (non-ductus-dependent lesions).
15.1.5 Clinical Features
The hemodynamic impact of a PDA in an otherwise normal cardiovascular system is determined by the magnitude of the shunt which depends on the ductal resistance and also on the pressure gradient between the aorta and the pulmonary artery. Small and restrictive PDAs usually cause little or no hemodynamic derangement and are asymptomatic. Rarely, bacterial endocarditis can occur. Moderate to large PDAs can result in pulmonary overcirculation and left heart volume overload. Chronic volume overload of the left heart can lead to onset of congestive heart failure in adulthood, usually starting in the third decade. In addition, long-standing left-to-right shunting and exposure of the pulmonary artery system to high pressures can lead to morphological changes in the pulmonary vasculature, resulting in increased pulmonary vascular resistance. When pulmonary vascular resistance exceeds systemic vascular resistance, ductal shunting reverses and becomes right to left (Eisenmenger physiology) [2].
15.1.6 Interventions
Ductus closure is usually indicated for patients who are symptomatic from significant left-to-right shunting through the PDA or who have left-to-right shunting resulting in left heart enlargement. The indications for closure of PDAs with small shunts, including those that are incidentally discovered, are less certain. Because endarteritis has been reported in patients with clinically silent PDAs, it has been advocated that any PDA should be routinely closed, especially given efficacy and safety of the percutaneous closure methods.
Transcatheter occlusion with coils or a duct occluder device has become the treatment of choice for most older children and adults with PDAs. Complete closure rates usually exceed 90–95 %. In infants, PDA closure may be successful with indomethacin treatment. If indomethacin treatment is unsuccessful, PDAs are then usually closed with an external clip or surgical ligation or division.
15.1.7 Cardiac Computed Tomography (CT) in the Evaluation of PDA
CT enables excellent anatomic depiction of PDAs [5]. When other imaging techniques do not provide sufficient data, CT can be used as an additional tool for diagnosis. Although the channel between the aorta and pulmonary artery can be seen on axial images, it is best seen on sagittal or oblique multiplanar and/or 3D volume-rendered reconstructions (Figs. 15.2 and 15.3). Demonstration of a jet of blood from the aorta into the pulmonary artery via the PDA confirms the patency of the shunt [6, 7]. Of importance, a small diverticulum at the site of the ductus should not be confused with a patent ductus (Fig. 15.4). Unlike a PDA, the diverticulum will not demonstrate a patent connection with the pulmonary artery.
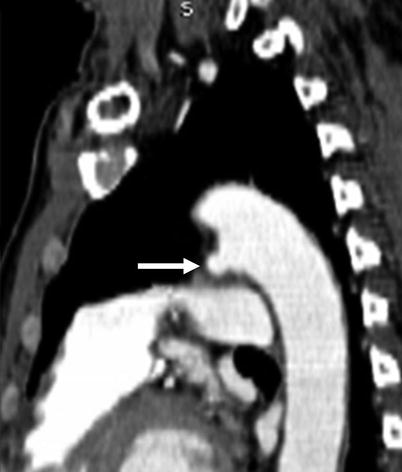
Fig. 15.4
Ductus diverticulum. The small diverticulum (arrow), which is a remnant of the patent ductus, does not connect with the pulmonary artery
Size and morphology of the ductus are important in planning closure procedures. The classification proposed by Krichenko et al. which divides PDA into five groups A–E (Table 15.1), related to the size of the PDA, presence or absence of constrictions, and, if present, the location of the constriction, has been useful to plan surgical or transcatheter closure procedure [8].
Type | Morphology |
|---|---|
A (conical) | Well-defined aortic ampulla and constriction near the pulmonary artery |
B (window) | Very large and very short |
C (tubular) | Without constrictions |
D (complex) | Multiple constrictions |
E (elongated) | Constriction remote from the anterior edge of trachea |
CT scans should be evaluated for the following: (a) location and size (length and width); (b) presence and extent of calcifications, constrictions, or aneurysms; (c) presence of vegetations; (d) size and morphology of the great arteries for the presence of pulmonary hypertension and dissection/rupture; (e) morphology, size, and function of the cardiac chambers, particularly the left atrium and left ventricle (which may be enlarged in patients with volume overload) and the right ventricle (which may be hypertrophied due to pulmonary hypertension and Eisenmenger syndrome); and (f) coexisting anomalies. See Table 15.2.
Table 15.2
CT assessment of PDA
Location, size, geometry, connections to great arteries, and relationship to neighboring structures |
Presence of constrictions, aneurysms, and wall calcifications |
Presence of vegetations suggestive of endocarditis |
Size and morphology of the great arteries including the presence of aorta and pulmonary artery dissection and/or spontaneous rupture |
Morphology, size, and function of the cardiac chambers |
Presence of coexisting anomalies |
Ductal aneurysm, which may occur prior to or after repair, aortic and/or pulmonary artery dissection, and/or spontaneous rupture are associated with this anomaly [9–14]. After closure procedures, CT may help to assess residual shunting, thrombus formation, and protrusion of transcatheter occluder devices into the arterial lumen [2].
15.2 Aortopulmonary Window
15.2.1 Definition
Aortopulmonary window (AP window) is a communication between the main pulmonary artery and the ascending aorta in the presence of two separate semilunar valves [15]. It is caused by incomplete fusion of the two embryonic conotruncal ridges that normally give rise to the aorta and the pulmonary artery [16].
15.2.2 Epidemiology
15.2.3 Variants
Based on the Mori classification, there are three variants of the AP window defect:
(a) type I or proximal communication located near the semilunar valves, (b) type II or distal connection involves the pulmonary bifurcation at the level of the right pulmonary artery, and (c) type III or a connection characterized by total absence of the aortopulmonary septum. Occasionally there may be an intermediate form that has features of types I and II (Figs. 15.5 and 15.6) [19, 20].
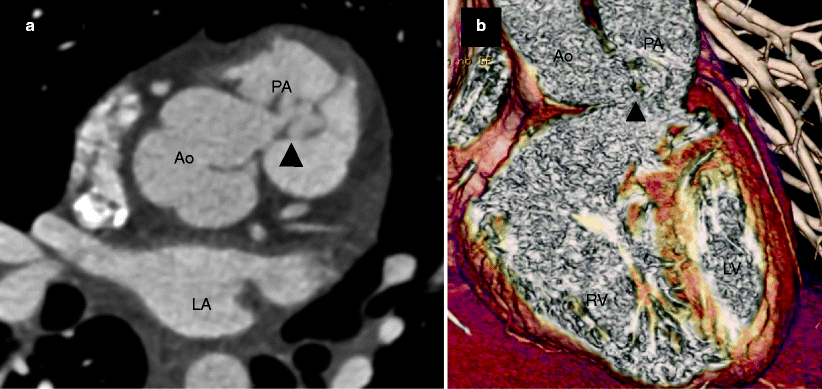
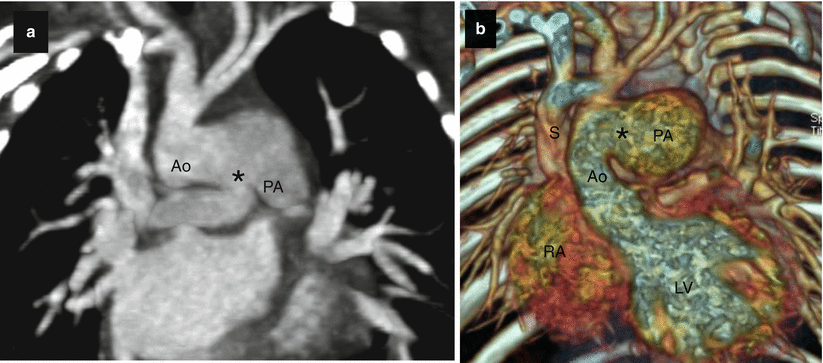
Fig. 15.5
Type I aortopulmonary window in a 20-year-old woman without prior surgical repair. Panel (a) is an axial scan, while panel (b) is a volume-rendered view demonstrating a communication (arrowheads) between the aorta (Ao) and main pulmonary artery (PA) at the level of the semilunar valves. LA left atrium, LV left ventricle, RV right ventricle (Images provided courtesy of Eric T. Kimura-Hayama MD, Mexico City, Mexico)
Fig. 15.6
Type II aortopulmonary window in a 3-month-old infant. Panel (a) is a two-dimensional tomogram and panel (b) is a volume-rendered, coronal reconstruction. Each shows a communication (asterisk) between the aorta (Ao) and pulmonary artery (PA) just above the plane of the semilunar valves. S superior vena cava, RA right ventricle, LV left ventricle (Images provided courtesy of Eric T. Kimura-Hayama MD, Mexico City, Mexico)
15.2.4 Associated Anomalies
About 50 % of AP window defects are associated with other cardiac abnormalities. The most common associated lesions are type A interruption of the aortic arch and aortic coarctation [15–17, 21, 22]. Other abnormalities include (a) anomalous origin of the right pulmonary artery from the right side of the ascending aorta, (b) anomalous origin of the coronary arteries, (c) ventricular septal defect, (d) tetralogy of Fallot, and (e) transposition of the great arteries (Table 15.3) [16].
The AP window needs to be distinguished from truncus arteriosus and hemitruncus, which also are characterized by communications between the aorta and pulmonary artery. In the AP window defect, there are separate aortic and pulmonary valves and the right and left pulmonary arteries arise from the main pulmonary artery. Truncus arteriosus has a single truncal valve. In hemitruncus, one pulmonary artery arises from the ascending aorta and the other from the right ventricle [23, 24].
15.2.5 Clinical Aspects of AP Window
Patients with AP window defects typically present in the neonatal period with heart failure and undergo cardiac surgery early in life [16]. The abnormality is rarely indentified later in adult life when dyspnea on exertion, cyanosis, and/or pulmonary hypertension develop. More often, adults present with postoperative complications, including residual or recurrent left-to-right shunt, obstruction of the ascending aorta or main or right pulmonary artery, or obstruction of a coronary artery. Surgical closure procedures include a pulmonary artery flap and pericardial patch. Small residual defects may be treated with a closure device implantation. Prognosis depends on the presence of coexisting anomalies. In general, survival is excellent in patients with isolated AP windows.
15.2.6 Cardiac Computed Tomography (CT) in the Assessment of AP Window
CT is a noninvasive tool to depict and characterize the AP window defect [25, 26]. In individuals with unrepaired AP windows, CT assessment should include (a) the presence, location, type, and size of the AP window; (b) size and morphology of the pulmonary artery and aorta, (c) size and function of the cardiac chambers; and (d) presence of coexisting cardiac abnormalities, including bronchial compression by dilated pulmonary arteries, which cannot be demonstrated by echocardiography (Table 15.4).
In patients who have undergone surgical repairs, the role of CT is to assess (a) the integrity of the patch or device closure and the presence of residual or recurrent shunting, (b) the course and size of the great vessels and pulmonary artery branches, and (c) the status of coexisting abnormalities (Table 15.5).
15.3 Aortic Coarctation and Interrupted Aortic Arch
15.3.1 Aortic Coarctation
Coarctation of the aorta is a congenital narrowing at the junction of the aortic arch and proximal descending aorta in the region of the ligamentum ductus arteriosus [1]. Demonstrating a male to female ratio as high as 2:1, it accounts for 5–8 % of all congenital heart defects with an incidence of 1 in 2,500 live births [27, 28].
Aortic coarctation is thought to be the result of a malformation of the aortic media, leading to a short-segment, shelflike posterior infolding of the aortic wall in the periductal region [28]. See Fig. 15.7. However, coarctation of the aorta may be a long segment and may also be associated with isthmus and arch hypoplasia [29–31]. Intrinsic aortic wall abnormalities, characterized by cystic changes in the aortic media associated with disruption of elastin and increased collagen deposition, predispose to dissection or rupture in the area of the coarctation as well as in the ascending or descending aorta [1].
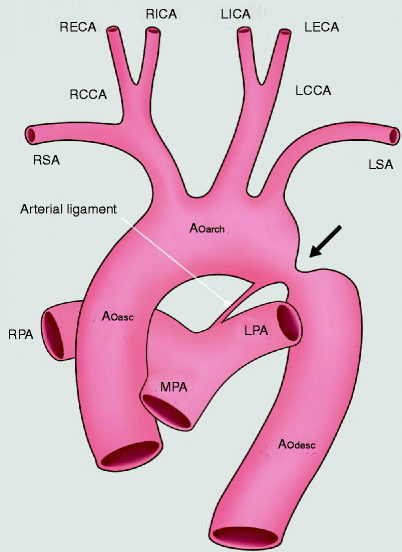
Fig. 15.7
An artist’s depiction of aortic coarctation. The coarctation (black arrow) is in the region of the embryologic ductus arteriosus at the junction of the transverse aortic arch and proximal descending aorta. Ao asc ascending aorta, Ao arch aortic arch, Ao desc descending aorta, LCCA left common carotid artery, RCCA right common carotid artery, LSA left subclavian artery, RSA right subclavian artery, MPA main pulmonary artery, LPA left pulmonary artery, RPA right pulmonary artery, RECA right external carotid artery, RICA right internal carotid artery, LECA left external carotid artery, LICA left internal carotid artery
Two major types of coarctation are recognized: (1) preductal (infantile) and (2) postductal (adult). In the preductal form of coarctation, the obstruction occurs immediately below the origin of the left subclavian artery at the insertion of the ductus arteriosus or ligamentum arteriosum (Fig. 15.8). This form of coarctation is associated with tubular hypoplasia of the transverse arch in addition to the focal constriction. The ductus arteriosus is usually patent and supplies blood to the descending aorta from the pulmonary artery.
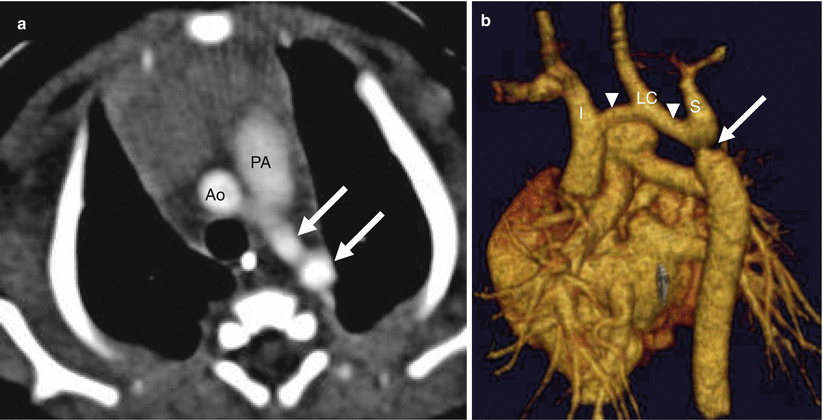
Fig. 15.8
Preductal coarctation. Panel (a) is an axial image showing the decreased caliber of the descending aorta (arrows) compared with the transverse aortic arch (Ao). PA pulmonary artery. Panel (b) is a sagittal 3D volume-rendered image demonstrating a focal constriction (arrow) of the aortic lumen below the left subclavian artery (S) and long-segment hypoplasia of the transverse arch (arrowheads). I innominate artery, LC left carotid artery
In the postductal form, coarctation occurs below the level of the obliterated ductus arteriosus and produces a focal, shelflike indentation of the posterior aortic wall [1, 27–33]. See Fig. 15.9. The areas of the aorta proximal and distal to the coarctation may be dilated. The high pressure gradient across the area of obstruction leads to the formation of systemic collateral circulation to maintain blood flow to the lower pressure distal aorta. Collateral pathways include (1) internal thoracic (mammary) arteries, (2) intercostal arteries, (3) thoracoacromial and descending scapular arteries, and (4) vertebral arteries (Figs. 15.9 and 15.10). Inferior rib notching of the third to sixth ribs by dilated intercostal arteries is another characteristic imaging finding.
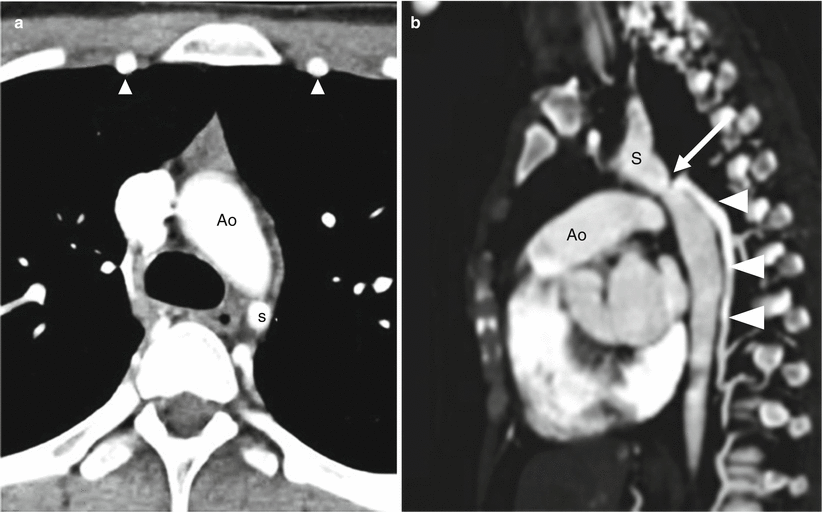
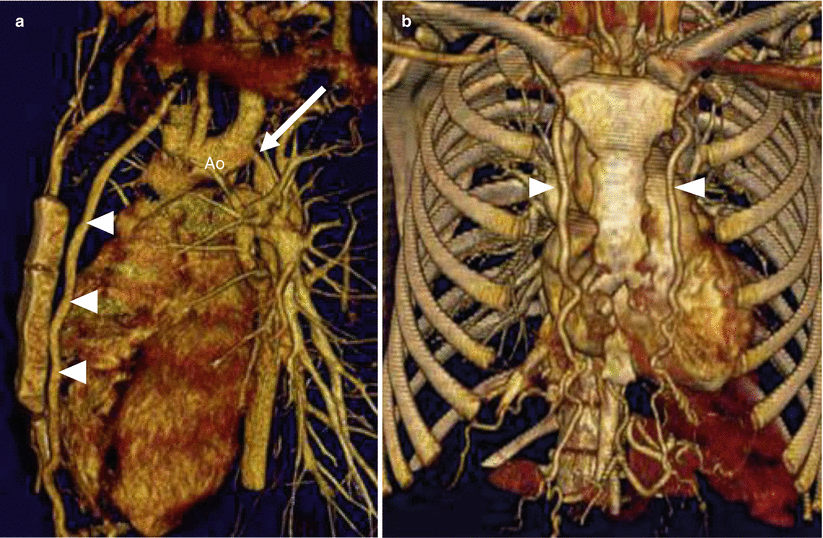
Fig. 15.9
Postductal aortic coarctation. Panel (a) an axial scan showing narrowing of the descending aorta (Ao) distal to the origin of the left subclavian artery (S). Also note the enlarged internal mammary arteries (arrowheads). Panel (b) sagittal image demonstrating the characteristic narrowing (arrow) of the aortic lumen at the level of the ligamentum arteriosum. Also note the large posterior collateral vessel (arrowheads) and the mildly dilated ascending aorta (Ao). S subclavian artery
Fig. 15.10
Aortic coarctation and collateral vessels. Panel (a) a sagittal scan shows the coarctation (arrow) and a large internal mammary artery (arrowheads). Panel (b) a coronal view confirms the enlarged internal mammary arteries (arrowheads). Ao aorta
Preductal coarctation is associated with ventricular septal defects, patent ductus arteriosus, and hypoplastic left heart. Postductal coarctation is frequently associated with bicuspid aortic valve (22–85 % of cases), parachute mitral valve, hypoplastic mitral valve, aortic arch hypoplasia, and ventricular septal defects [1]. Bicuspid aortic valve may be complicated by aortic stenosis or regurgitation or both. Coarctation may also coexist with complex congenital heart diseases including transposition of the great arteries, double-inlet ventricle, double-outlet right ventricle, tricuspid atresia, and hypoplastic left heart syndrome [28].
Syndromes associated with aortic coarctation include Turner syndrome (15–20 % incidence of coarctation) and Shone’s complex (left ventricular outflow tract obstruction and parachute mitral valve) [34].
Noncardiac associations include intracranial aneurysms (up to 10 % of coarctation cases) and spinal stenosis [1, 28, 32, 33, 35].
The clinical presentation and age at diagnosis of aortic coarctation are dependent on the severity of the obstruction and presence of other anomalies. The preductal form presents in infancy with congestive heart failure. Postductal coarctation can present in children or adults and may come to clinical attention because of a left sternal border systolic ejection murmur, systemic arterial hypertension in the upper extremities, and/or decreased lower extremity pulses [1, 27–31, 36]. Chronic hypertension and cerebrovascular accidents are late findings [36].
Untreated coarctation has a poor prognosis. Approximately 60 % of untreated infants with high-grade coarctation and 90 % with complicated coarctation die in the first year of life [28, 37]. In untreated patients, the mortality rate is reported to be 25, 50, 75, and 92 % at 20, 32, 46, and 60 years of age, respectively [38]. Surgery has substantially improved the long-term outcomes in infants with coarctation, although the survival rate in this group is still lower than that in an older population [38]. Coarctation in untreated adults may be asymptomatic due to the formation of systemic collateral vessels that reduce the gradient across the coarctation site masking the obstruction [1, 27, 28].
Treatment for coarctation consists of either open surgical repair or catheter interventions, including angioplasty and stent implantation. Resection and end-to-end anastomosis is the procedure of choice in neonates and infants with coarctation, although balloon angioplasty has been introduced as the bridge to surgery in critically ill subjects [39]. In adults, aortic coarctation can be treated percutaneously or with surgery [40, 41]. Surgical techniques include (a) resection and end-to-end anastomosis, (b) resection and extended end-to-end anastomosis, (c) prosthetic arch aortoplasty, (d) subclavian flap aortoplasty, (e) interposition of a (tube) graft, and (f) bypass tube grafting [28, 29]. See Chap. 32 for a discussion of aortic coarctation surgical repairs.
15.3.2 Interrupted Aortic Arch
Interrupted aortic arch is a form of coarctation characterized by the lack of luminal continuity between ascending and descending aorta (Fig. 15.11). The condition is rare, accounting for 1 % to all 1.5 % of congenital heart disease [28, 42]. The Celoria–Patton classification identifies three types of interrupted arch based on the site of interruption. In type A, the interruption is distal to the left subclavian. Type B interruptions occur between the left carotid and left subclavian artery, and type C interruptions reside between the brachiocephalic trunk and left carotid artery [42]. In patients with an interrupted aortic arch, blood flow to the descending aorta is provided by an obligatory persistent arterial duct (Fig. 15.11).
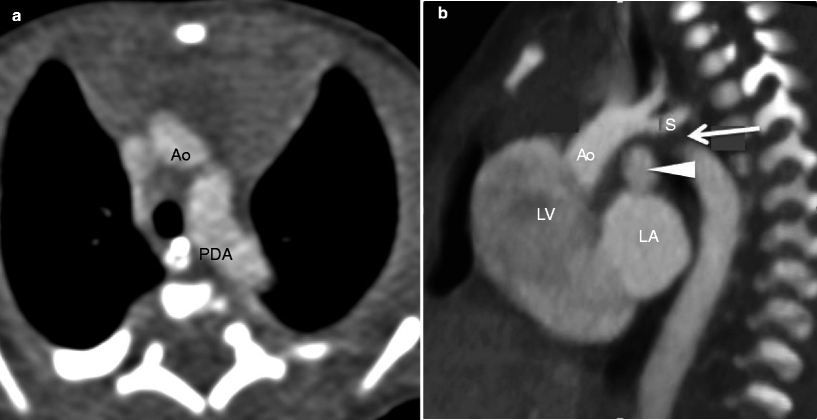
Fig. 15.11
Interrupted aortic arch in a neonate. Panel (a) an axial section demonstrates a normal caliber transverse aortic arch (Ao). The vessel inferior to the arch is a large dilated patent ductus arteriosus (PDA), not to be confused with the descending aorta. Panel (b) a sagittal reconstruction shows interruption of the transverse arch (arrow) just below the origin of the left subclavian artery (S) and the large patent ductus arteriosus (arrowhead) which supplies the distal aorta. The ascending aorta is normal caliber. Ao ascending aorta, LV left ventricle, LA left atrium
This anomaly is commonly associated with ventricular septal defects, bicuspid aortic valves, left ventricular outflow tract obstruction, and aberrant right subclavian artery originating from the descending aorta. It also may be associated with complex heart lesions including common arterial trunk, aortopulmonary window, double-outlet right ventricle, and transposition of great arteries [42]. Type B interruption has been associated with the DiGeorge syndrome (hypocalcemia and T-cell defects due to thymic hypoplasia) [42]. Patients with interrupted aortic arch present in the neonatal period with respiratory distress, cyanosis, and congestive heart failure.
Table 15.3
Coexisting anomalies in aortopulmonary window
Coarctation of aorta or interrupted aortic arch |
Abnormal origin of the right pulmonary artery from ascending aorta |
Coronary artery anomalies |
Ventricular septal defect |
Tetralogy of Fallot |
Transposition of great arteries |
Table 15.4
Computed tomography assessment in untreated patients with aortopulmonary window
Presence, location, type, and size of aortopulmonary window |
Morphology, size, and course of great arteries |
Morphology, size, geometry, and systolic function of both ventricles with outflow tracts |
Morphology and size of atria |
Presence of coexisting anomalies |
Table 15.5
Computed tomography assessment in treated patients with aortopulmonary window
Integrity of aortopulmonary window closure patch/device (presence of residual or recurrent shunting) |
Morphology, size, course, and obstruction of great arteries |
Integrity of repair of coexisting anomalies |
Morphology, size, and geometry of both ventricles with their outflow tracts, including systolic function of the ventricles |
Morphology and size of atria |
Surgical techniques for repair of the interrupted arch include (a) direct anastomosis after mobilization of both the ascending and descending aorta, (b) direct anastomosis with patch augmentation, and (c) interposition of a conduit. If necessary, the closure of ventricular septal defect and resection of the aortic stenosis is performed [43]. Mortality with these techniques ranges from 15 to 20 % [44]. The abnormality can be identified later in adult life if stenosis or significant collateral circulation develops [45].
15.3.3 Aortic Pseudocoarctation
Aortic pseudocoarctation is an unusual anomaly characterized by buckling or kinking of a tortuous aortic arch at the level of the ligamentum arteriosus (Fig. 15.12). It is thought to be the result of incomplete fusion of the third and seventh aortic dorsal segments. Although there may be some mild luminal narrowing and turbulence, the essential feature of pseudocoarctation is the absence of a hemodynamically significant pressure gradient. Therefore, there is no collateral vessel formation. This anomaly is usually asymptomatic and discovered incidentally on imaging studies and needs to be differentiated from a true aortic coarctation. Pseudocoarctation need not be treated.
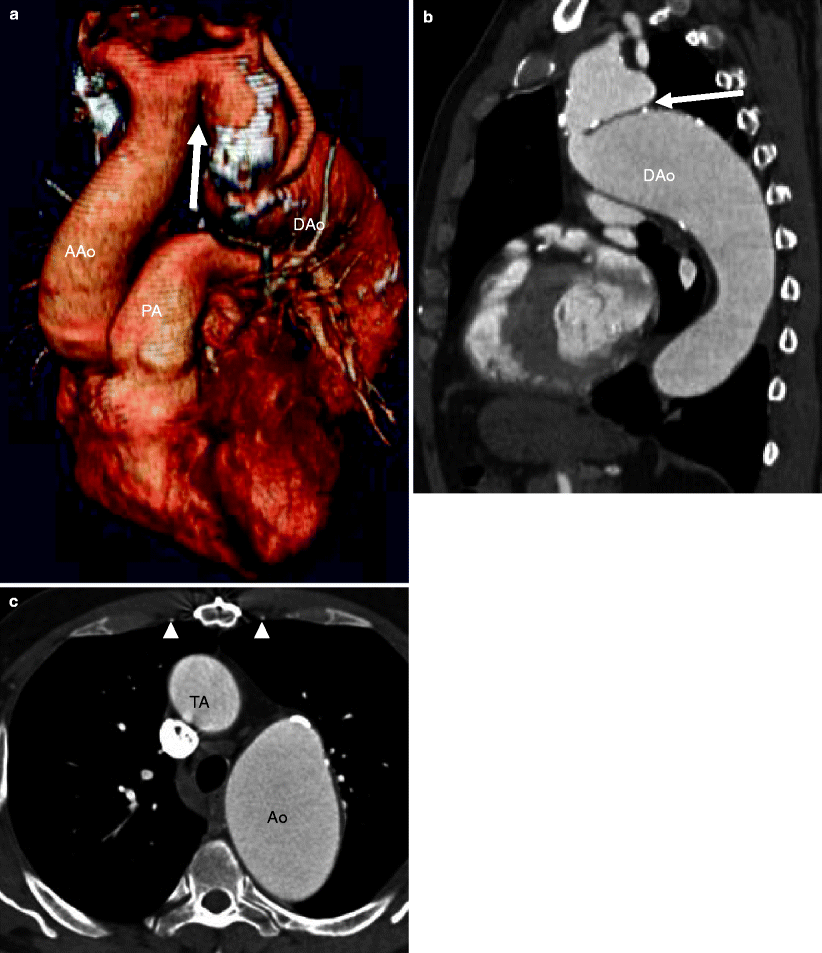
Fig. 15.12
Pseudocoarctation. Panel (a) (3D sagittal) and panel (b) (oblique maximum intensity projection) reconstructions demonstrating the pronounced buckling of a tortuous aorta at the level of the ligamentum arteriosus (arrows). The “shelflike” fold is due to the buckling of the aorta. Note the absence of collateral vessel formation. Panel (c) an axial scan highlights that the internal mammary arteries (arrowheads) are normal caliber. TA transverse aortic arch, AAo ascending aorta, Dao descending aorta, PA pulmonary artery
15.3.4 Cardiac Computed Tomography (CT) in the Evaluation of Aortic Coarctation and Interrupted Aortic Arch
In the untreated coarctation and interrupted aortic arch, CT enables a detailed assessment of anatomy including the presence, site, morphology, and extent of luminal narrowing at the coarctation site as well as at the hypoplastic segment of the aortic arch and isthmus. Additionally, associated aortic dilatation, aneurysm formation, or aortic dissection may be identified. Luminal narrowing of the aortic arch and its branches can be seen. Collateral circulation may be identified. Finally, accompanying anomalies such as bicuspid aortic valve, mitral valve abnormalities, ventricular septal defects, and other congenital heart diseases should be identified [5, 46, 47]. However, CT cannot provide functional data such as the gradient across the coarctation.
In aortic coarctation, CT may identify collateral flow which is important in surgical planning. The aorta may be cross-clamped during surgical repair if collateral flow is sufficient for lower body perfusion. If collateral formation is inadequate, use of cardiopulmonary bypass for perfusion of the lower body may be indicated. Reconstructed images in sagittal and parasagittal planes best show the location and extent of coarctation. Coronal and sagittal reconstructions may help delineate rib notching and or collateral vessel formation.
In the post-intervention patient, the goals of CT imaging include evaluation of residual or recurrent stenosis(es), identification of potential endovascular stent-related complications (leaks, stent migration, stent fracture, in-stent thrombus, and aortic dissection), assessment of aortic dilatation, aneurysm formation and dissection, delineation of the aortic branch artery anatomy, assessment for collateral vessel formation, and characterization of any coexisting anomalies [48–50].
Since patients with aortic coarctation may experience accelerated atherosclerosis and premature coronary artery disease, evaluation of the coronary arteries is critical. Therefore, any CT evaluation should provide a noninvasive assessment of the presence, severity, and extent of coronary atherosclerosis in these patients. See Tables 15.6 and 15.7.
Table 15.6
CT assessment in patients with coarctation of the aorta
Presence, site, morphology, and extent of luminal narrowing and or the hypoplastic segment |
Presence, site, morphology, and extent of aortic dilatation and aneurysm formation of dissection |
Presence, site, morphology, and extent of luminal narrowing of the aortic arch and branch vessels |
Collateral circulation |
Accompanying anomalies, especially bicuspid aortic valve and its complications |
Other associated congenital heart disease |
Presence, extent, and severity of associated coronary atherosclerosis |
Table 15.7
CT assessment in postoperative patients with aortic coarctation
Presence, site, morphology, extent of residual luminal narrowing and/or recoarctation |
Endovascular stent-related complications, including leak, migration, fracture, thrombosis presence, site morphology, and extent of aortic dilatation, aneurysm, or dissection |
Anatomy and patency of aortic arch branch arteries |
Presence and extent of collateral vessel formation |
Coexisting anomalies including bicuspid aortic valve with its complications other associated congenital heart disease |
Presence, extent, and severity of associated coronary atherosclerosis |
15.4 Aortic Arch Anomalies
Aortic arch anomalies represent a wide spectrum of symptomatic and asymptomatic congenital malformations that affect both the aortic arch and its branches.
Knowledge of the embryology of the great arteries is helpful in understanding the variations in the number, size, and position of the thoracic great vessels.
The normal left aortic arch and its branches develop from paired pharyngeal arches which form during the 4th and 5th weeks of development and undergo a process of orderly regression. There are six potential arches in the human embryo that are numbered craniocaudally as I, II, III, IV, V, and VI. Arch V may never form or incompletely forms and then regresses. Arches III, IV, and VI develop into the great arteries (Figs. 15.13 and 15.14). Arch III forms the common carotid arteries and the first part of the internal carotid arteries. Right arch IV forms the most proximal segment of the right subclavian artery, and left arch IV develops into the segment of the aortic arch between the left common carotid and the left subclavian arteries. Right arch VI creates the right pulmonary artery, and the left arch VI becomes the left pulmonary artery and the ductus arteriosus.
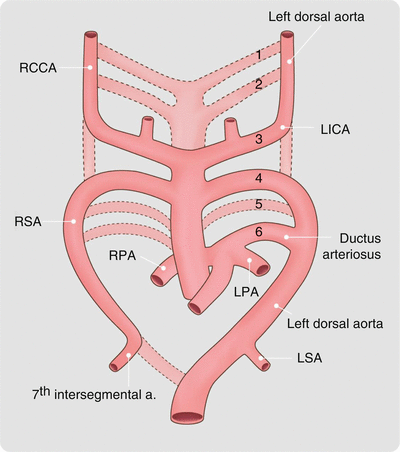
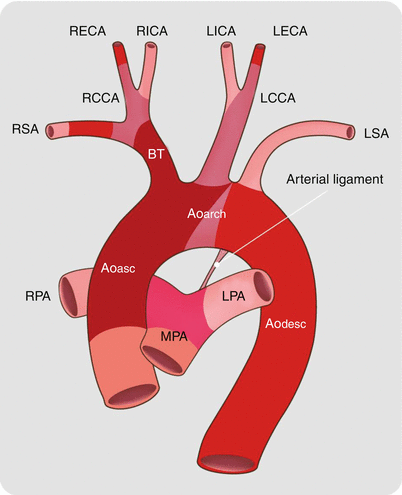
Fig. 15.13
Normal embryologic development of the great arteries. At 6 weeks, the first and second pairs of branchial arches have virtually disappeared. The third arch forms the common carotid arteries. The right fourth arch becomes the proximal part of the right subclavian artery and the left fourth arch becomes part of the normal aortic arch. The distal parts of the subclavian arteries develop from the seventh intersegmental arteries. The fifth arch either never forms or involutes early. The sixth pair of arches forms the pulmonary arteries bilaterally. The left sixth arch also gives rise to the ductus arteriosus. Arches are numbered 1–6. RCCA right common carotid artery, LICA left internal carotid artery, RSA right subclavian artery, RPA right pulmonary artery, LSA left subclavian artery, LPA left pulmonary artery
Fig. 15.14
The normal adult aortic arch. The right subclavian and carotid arteries join to form the right brachiocephalic artery. The left subclavian and carotid arteries arise separately from the aorta. Aoasc ascending aorta, Aoarch aortic arch, Aodesc descending aorta, MPA main pulmonary artery, RPA right pulmonary artery, LPA left pulmonary artery, BT brachiocephalic trunk, RSA right subclavian artery, RCCA right common carotid artery, RECA right external carotid artery, RICA right internal carotid artery, LCCA left common carotid artery, LICA left internal carotid artery, LECA left external carotid artery, LSA left subclavian artery
In the 5th week, the two aortas fuse caudally below T4 to form a single aorta in the distal thorax and abdomen. Persistence of a segment of arch that should have regressed or regression of a segment of arch that should have persisted accounts for the development of most arch anomalies.
Arch anomalies can be subdivided into anomalies of position and arch branching and anomalies of number (i.e., supernumerary arches). The abnormalities of branching and position include left aortic arch with aberrant left subclavian artery, right aortic arch with mirror-image branching, right aortic arch with aberrant left subclavian artery, and cervical aortic arch. Supernumerary arches include double aortic arch and persistent fifth aortic arch.
Aortic arch anomalies may produce vascular rings that surround the thoracic anatomy (trachea and esophagus). The primary symptomatology associated with vascular rings relates to the structures that they encircle and may include dysphagia, odynophagia, wheezing, and shortness of breath with and without exertion. Arch anomalies that completely encircle the trachea and bronchi are referred to as “vascular rings.” Vascular rings are more likely to be symptomatic than arch anomalies that only partially surround adjacent structures. The majority of symptomatic arch anomalies are diagnosed in infants; however, adults with vascular rings may be symptomatic.
15.4.1 Left Aortic Arch with Aberrant Right Subclavian Artery
The left aortic arch with an aberrant right subclavian artery is the most common congenital abnormality of the aortic arch vessels and occurs in about 0.5–2 % of the general population [51, 52].
Normally, the right subclavian artery is a continuation of the brachiocephalic artery which is the first major aortic arch branch vessel. In left aortic arch with aberrant right subclavian artery, the right subclavian artery is the last branch off the aortic arch. In this anomaly, the proximal to distal order of branching is as follows: right common carotid, left common carotid, left subclavian, and then the aberrant right subclavian (Figs. 15.15 and 15.16). The anomalous artery courses cephalad from left to right, crossing behind the trachea and esophagus to reach the right arm. Dilatation of the origin of the aberrant vessel, termed a diverticulum of Kommerell, is common (Fig. 15.17). The diverticulum of Kommerell itself may give rise to right subclavian artery. Atherosclerotic changes and intramural thrombus formation are known to occur within this diverticulum.
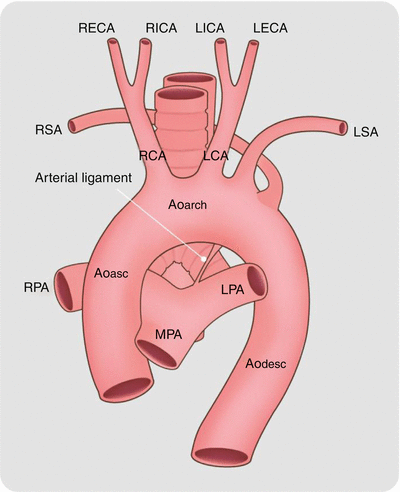
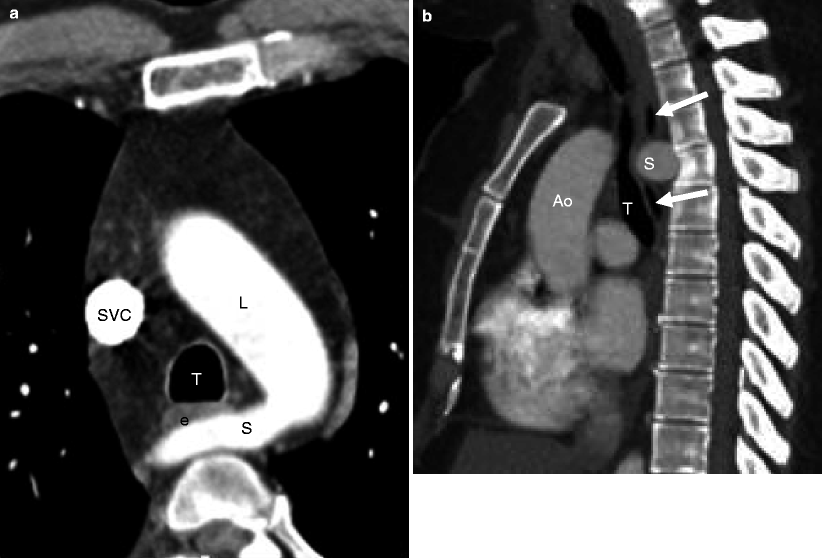
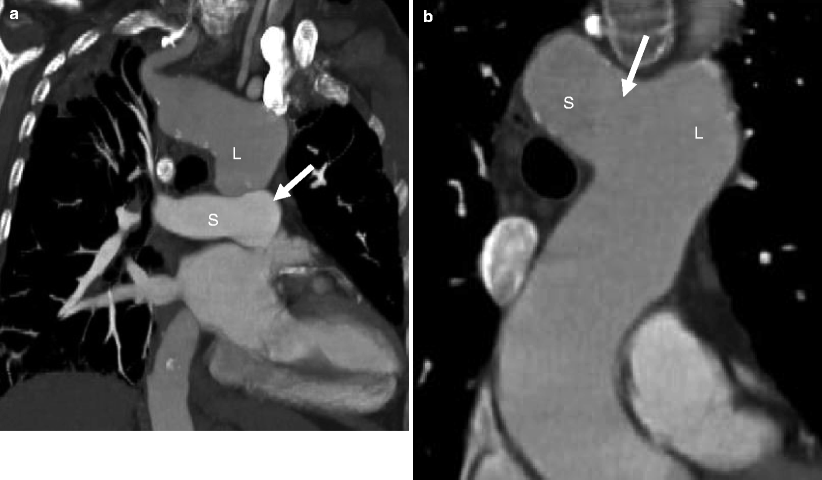
Fig. 15.15
Left aortic arch with aberrant right subclavian artery. Note the right subclavian artery (RSA) is the last branch off the arch. Ao asc ascending aorta, Ao arch aortic arch, Ao desc descending aorta, LPA left pulmonary artery, LSA left subclavian artery, LCA left carotid artery, MPA main pulmonary artery, RPA right pulmonary artery, RCA right carotid artery, Arrow ligamentum ductus arteriosum, RECA right external carotid artery, RICA right internal carotid artery, LICA left internal carotid artery, LECA left external carotid artery
Fig. 15.16
Left aortic arch with an aberrant right subclavian artery. Panel (a) is an axial image showing a left aortic arch (L) and an aberrant right subclavian artery (S) coursing posterior to the esophagus (e) and trachea (T). Panel (b) is a sagittal image demonstrating the right subclavian artery (S) coursing posteriorly and compressing both the esophagus (arrows) and trachea (T). Ao aorta, SVC superior vena cava
Fig. 15.17
Left arch with aberrant right subclavian artery and diverticulum of Kommerell. Panels (a) and (b) are coronal views showing the left aortic arch (L) and an aberrant right subclavian artery (S), which is focally enlarged at its origin. This focal enlargement is termed a diverticulum of Kommerell (arrow)
Left aortic arch with aberrant right subclavian artery is associated with a left ductus arteriosus (ductus arteriosum ligament) which completes the vascular ring. This malformation may be associated with other congenital heart diseases, most frequently tetralogy of Fallot and ventricular septal defects [50]. Its incidence is also increased in trisomy 21 syndrome [53].
This anomaly is usually asymptomatic and found incidentally on imaging examinations. However, if the aberrant subclavian artery is ectatic, it may compress the esophagus resulting in dysphagia (dysphagia lusoria). It may occasionally also result in airway compression which may result in wheezing and shortness of breath.
15.4.2 Rare Left Aortic Arch Anomalies
Left aortic arch with right descending aorta and a right arterial duct/ligament describes an arch anomaly in which the left-sided aortic arch takes a retroesophageal course and, from proximal to distal, gives rise first to the right carotid artery, followed by left carotid artery, left subclavian artery, and finally the right subclavian artery [51]. A vascular ring is completed by the right arterial duct/ligament.
Left aortic arch with isolation of the subclavian artery depicts an arch anomaly where the left subclavian artery originates exclusively from the pulmonary artery via an arterial duct or ligament. The subclavian artery is supplied by the pulmonary artery if the ductus arteriosus is patent. When arterial duct closes, the subclavian artery is supplied by retrograde flow from the vertebral artery.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


