Abstract
Autosomal recessive polycystic kidney disease, also called infantile polycystic kidney disease, is a chronic, progressive condition that causes cystic dilatation of the renal collecting ducts and congenital hepatic fibrosis. It is caused by mutations in the PKHD1 gene and has a wide spectrum of phenotypic variability. Approximately 30% of cases manifest prenatally or in the neonatal period, typically with a 50% mortality rate, but the remainder have milder and more variable disease. The characteristic prenatal sonographic finding is massive, symmetrically enlarged and echogenic kidneys that fill and distend the fetal abdomen. With advancing gestation, tiny cysts may be visible. These classic cases are associated with Potter sequence—lack of amniotic fluid resulting in pulmonary hypoplasia that is often lethal. More challenging are cases with milder phenotype because the degree of renal enlargement is less impressive and echogenicity may be apparent only after mid-gestation. The differential diagnosis for these findings includes several genetic syndromes as well as a normal variant, and amniocentesis should be offered. Molecular genetic testing may be helpful in affected families.
Keywords
polycystic kidney, phenotypic variability, variable expressivity, hepatic fibrosis
Introduction
Autosomal recessive polycystic kidney disease (ARPKD) is a chronic, progressive condition that affects the kidneys and liver, causing cystic dilatation of the renal collecting ducts and congenital hepatic fibrosis (CHF), or Caroli disease. ARPKD is also called infantile polycystic kidney disease and ARPKD/CHF. ARPKD is caused by mutations of a large, complex gene, PKHD1, and it has an unusually wide spectrum of phenotypic variability. Diagnosis is made prenatally or soon after birth in approximately 30% of patients, and neonatal mortality from pulmonary hypoplasia occurs in approximately 15% of cases—up to half of cases with prenatal diagnosis. Among children who survive infancy, morbidity is determined by the degree of renal insufficiency, with current 5-year and 10-year survival rates between 80% and 90%. Some individuals do not come to medical attention until later in childhood or adulthood and have both renal and hepatic manifestations, including early-onset hypertension, renal failure, portal hypertension, and recurrent cholangitis. The varying phenotypic manifestations of ARPKD can pose unique challenges from the standpoint of prenatal diagnosis, particularly in absence of an informative family history.
Disorder
Definition
Infantile polycystic kidney disease is an autosomal recessive disease that causes cystic dilatation of the renal collecting ducts and CHF.
Prevalence and Epidemiology
The prevalence of ARPKD is estimated to be 1 : 20,000 births. Inheritance is autosomal recessive, with complete penetrance but variable expressivity even within a family. Carrier frequency of a disease-causing PKHD1 mutation is estimated to be 1 : 70 in the general population. Family members of affected individuals should be counseled about inheritance patterns and recurrence risk.
Etiology and Pathophysiology
Historically, ARPKD was divided into four subtypes, based on timing of presentation and degree of renal and hepatic involvement: perinatal, neonatal, infantile, and juvenile. It is now viewed as a spectrum of disease. Renal involvement is characterized by dilatation and elongation of the cortical collecting ducts, resulting in a uniform distribution of radially arranged fusiform cysts. The outer renal cortex is spared because it contains no tubules. In the liver, proliferation and dilatation of portal bile ducts are responsible for development of periportal fibrosis. ARPKD is caused by mutations of the PKHD1 gene, which is located on the short arm of chromosome 6. PKHD1 is one of the largest human genes, possessing a complex splicing pattern that results in multiple transcripts, the largest of which is fibrocystin/polyductin. This gene product is expressed in the kidney and, to a lesser extent, in the liver, and it is believed to play a role in regulation of cell proliferation, adhesion, and repulsion.
Molecular genetic testing is available for affected families. PKHD1 mutation screening using denaturing high-performance liquid chromatography has identified mutations in more than 75% of cases, including 85% of patients with perinatal or neonatal demise. However, because most affected children are compound heterozygotes (inheriting a different mutation from each parent) and because most mutations are unique to individual families, genotype-phenotype correlations pose a challenge. In general, inheritance of two truncating mutations is more strongly associated with perinatal mortality, whereas those with missense mutations tend to display a milder phenotype. Prenatal diagnosis is available using single gene molecular genetic analysis, as is preimplantation genetic diagnosis. Indirect, haplotype-based linkage analysis is no longer the preferred method because of the possibility of misdiagnosis.
Manifestations of Disease
Clinical Presentation
The clinical presentation is highly variable. Diagnosis may be made in affected individuals before birth, in the neonatal period, in childhood, or in young adulthood. Manifestations may include pulmonary hypoplasia, early-onset hypertension, renal insufficiency, portal hypertension, esophageal varices, and recurrent cholangitis. Approximately 50% of patients require dialysis by age 20. Prenatally, the diagnosis is suspected based on ultrasound (US) findings and may be confirmed in an informative family by the identification of disease-causing mutations.
Imaging Technique and Findings
Ultrasound.
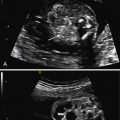
The characteristic US finding is massive, symmetrically enlarged, echogenic kidneys, which fill and distend the fetal abdomen, measuring between 4 and 15 standard deviations above the mean for gestational age. Normal corticomedullary differentiation is not visible. In some cases, these findings may be apparent in the first trimester ( Fig. 16.1 ). When ARPKD manifests early in gestation, amniotic fluid volume is usually severely decreased, with no urine visible in the bladder ( Fig. 16.2 ). As with other causes of severe, prolonged oligohydramnios, there is significant risk for pulmonary hypoplasia secondary to Potter sequence (see Chapter 10 ). With advancing gestation, the appearance of the kidney becomes more inhomogeneous, and tiny cysts may be visible ( Fig. 16.3 ).



In many cases, the diagnosis of ARPKD is not so straightforward. Another presentation involves kidneys that are echogenic but only mildly enlarged, measuring between 2 and 4 standard deviations above the mean, with preserved amniotic fluid volume ( Fig. 16.4 ). The kidneys may not appear noticeably abnormal until after midgestation. In the absence of an informative family history, counseling for such cases is problematic. Although the differential diagnosis includes ARPKD, it also includes genetic syndromes such as Bardet-Biedl syndrome and glutaric aciduria type II, aneuploidy such as trisomy 13 ( Fig. 16.5 ), and especially, normal kidneys. A careful family history, diligent search for other anomalies, and consideration of amniocentesis are important.


When the fetal kidneys are mildly enlarged and echogenic, and amniotic fluid volume is normal, the differential diagnosis also includes autosomal dominant polycystic kidney disease (ADPKD) ( Fig. 16.6 ). ADPKD usually has no prenatal manifestations; patients typically present in the fourth decade of life. However, ADPKD is much more common than ARPKD, occurring in 1 : 800 births rather than 1 : 20,000. In the absence of a family history or other fetal abnormalities, consideration may be given to US evaluation of the parents’ kidneys ( Fig. 16.7 ).

Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


