of head trauma and assess blood flow in cases of cerebrovascular disease. A sonographic finding or suspected lesion can be further characterized with computed tomography (CT) or magnetic resonance imaging (MRI) when the patient is more stable.
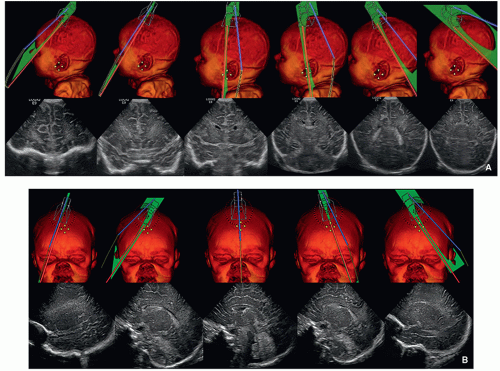 FIGURE 2.1 A: Positions of the transducer for coronal ultrasound images through the brain. B: Positions of the transducer for sagittal ultrasound images through the brain. |
tectum of the midbrain, and fourth ventricle can be identified on this midline image.
factors necessitate use of sedation or general anesthesia in a proportion of young pediatric patients. Various techniques have been used to reduce the need for sedation, including mock MRI, which acclimatizes the child to the MRI scanner, and technologies that decrease acquisition times, such as multichannel head coils, parallel imaging, and motion compensation techniques. In order to ensure that the information required to answer the clinical question is acquired in the shortest possible time before lack of patient cooperation becomes problematic, it is important to acquire the most important sequences at the start of the examination and actively monitor the MRI exam in a pediatric patient.
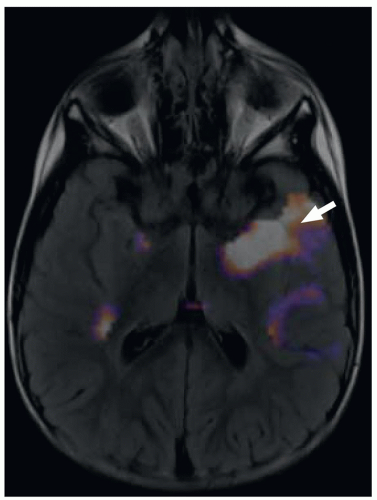 FIGURE 2.2 A 3-year-old boy with temporal lobe epilepsy. Axial subtraction ictal SPECT coregistered to MRI (SISCOM) image demonstrates a left temporal lobe epileptogenic focus (arrow). |
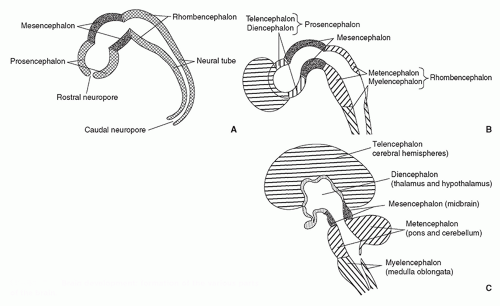 FIGURE 2.3 Brain development: formation of the various parts of the brain. |
they differentiate to form the normal six-layered cortex (Fig. 2.4).
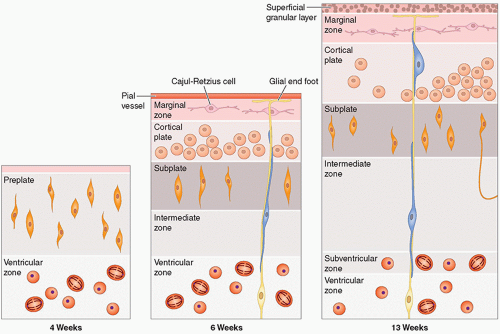 FIGURE 2.4 Cortical development. |
are affected by a number of factors, including changes in the structure and chemical composition of the axon and the surrounding myelin and the degree of axonal ensheathment by myelin. Most authors believe that signal changes on T1-weighted MR images correspond with the increase in the certain lipids occurring during myelin formation. The changes on T2-weighted MR images can be explained by myelin sheath maturation and decrease in the water content of the white matter indicated in vitro by thickening and tightening of the spiral of myelin around the axon.
TABLE 2.1 Milestones of Myelination | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
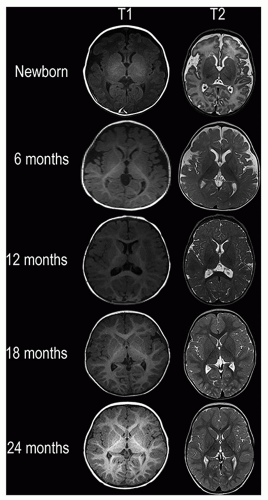 FIGURE 2.5 Summary of the stages of myelination from the newborn period to 24 months of age on T1- and T2-weighted MR images. |
TABLE 2.2 Classification of Congenital Brain Malformations | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
lamination of the cortex. The overfolding is usually microscopic, and the abnormal lamination either unlayered or four-layered in most described cases. Polymicrogyria is most commonly seen in the perisylvian cortex, but almost all cortical regions can be involved. Polymicrogyria is a highly heterogeneous disorder in terms of its pathogenesis, distribution, pathologic appearance, and clinical and imaging features. The clinical presentation is varied, and patients present at all ages from the neonatal period until late adulthood.
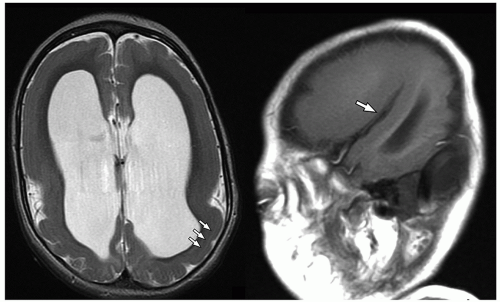 FIGURE 2.6 Incomplete lissencephaly (pachygyria) in a 3-year-old boy. Axial T2-weighted MR image (on the left) shows lissencephaly characterized by broad gyri and shallow sulci more pronounced in the frontal and temporal lobes. Note the T2 hyperintense cell-sparse zone in the parietal lobes (white arrows). Sagittal T1-weighted MR image (on the right) shows a vertically oriented Sylvian fissure (white arrow). |
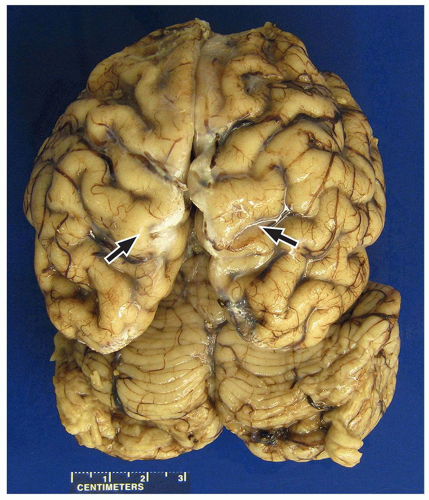 FIGURE 2.7 Pachygyria in an 11-year-old girl with microcephaly. The cerebral cortex shows pachygyria, characterized by a paucity of gyri (black arrows). |
On MRI, polymicrogyria is characterized by an irregular cortical surface and “stippled” gray-white junction with regions of apparent cortical thickening (Figs. 2.8 and 2.9).
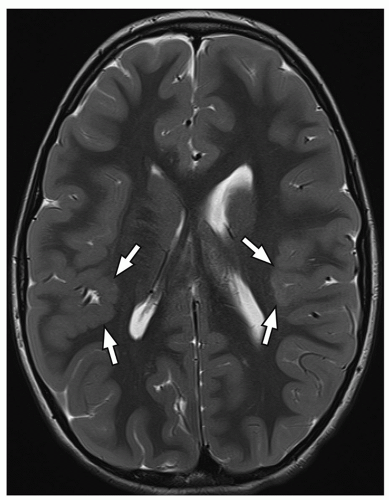 FIGURE 2.8 Bilateral perisylvian polymicrogyria. Axial T2-weighted MR image shows the irregular nodular cortical-white matter junction (arrows) in the bilateral perisylvian regions, right more than left. |
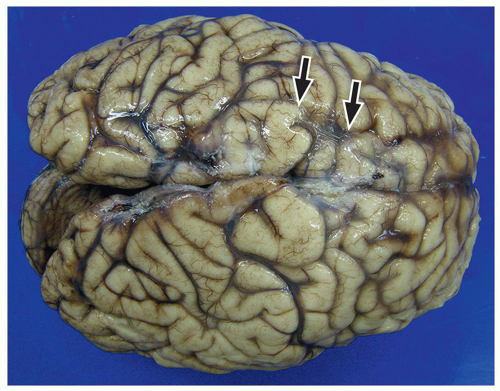 FIGURE 2.9 Polymicrogyria in a 13-year-old boy with holoprosencephaly. Superior aspect of the brain shows polymicrogyria of the frontal (right) and anterior parietal lobes (arrows). |
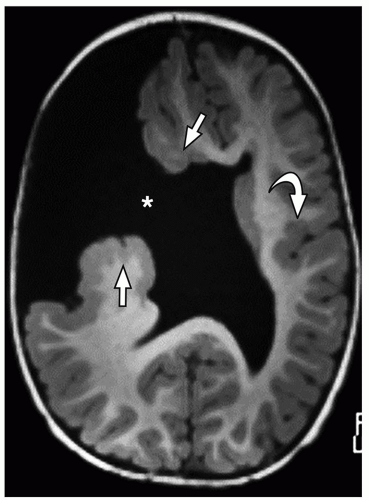 FIGURE 2.10 Open-lip schizencephaly. Axial MPRAGE (magnetization-prepared rapid acquisition gradient-echo) MR image demonstrates a large right-sided full-thickness cleft (asterisk), lined by abnormal polymicrogyric gray matter (straight arrows), that communicates with the lateral ventricle. Also, note absence of the septum pellucidum in this child with septooptic dysplasia and polymicrogyria (curved arrow) without schizencephaly in the left cerebral hemisphere. |
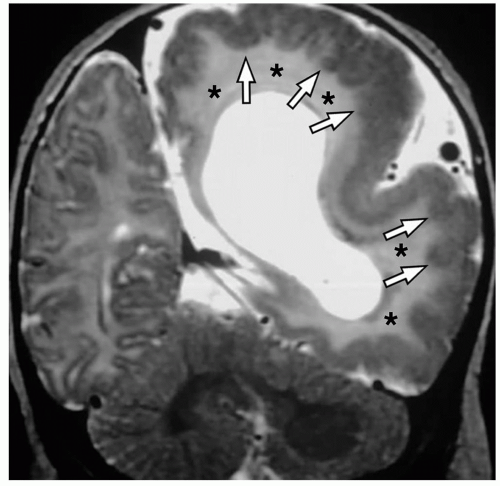 FIGURE 2.11 Hemimegalencephaly. Coronal T2-weighted MR image shows the abnormal dysplastic-appearing left hemisphere with an abnormal gyral pattern, cortical thickening, white matter hyperintensity (asterisks), and polymicrogyria (arrows). |
ependyma to the pia. The overlying cortex is thin, and the underlying ventricle often appears distorted. These foci follow gray matter on all sequences, do not demonstrate edema, and do not enhance, which differentiates them from glioneuronal tumors like gangliocytomas.
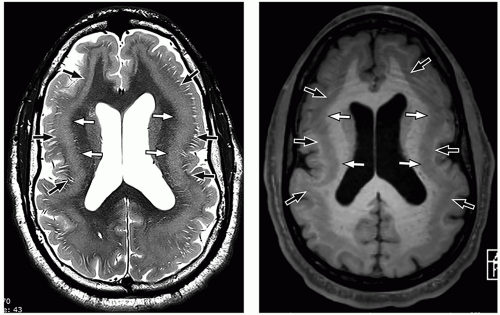 FIGURE 2.12 Subcortical band heterotopia. Axial T2-weighted (on the left) and axial SPGR (spoiled gradient recalled) (on the right) MR images demonstrate the double cortex appearance caused by an inner band of heterotopic gray matter (white arrows) separated from the outer cortex by a thin zone of normal white matter (black arrows). |
the abnormalities may also involve eloquent areas, and resection may not be an option in these cases. Therefore, other diagnostic imaging techniques such as FDG-PET, magnetoencephalography (MEG), DTI, and intracranial electroencephalography (EEG) are widely used to establish the diagnosis and plan management.
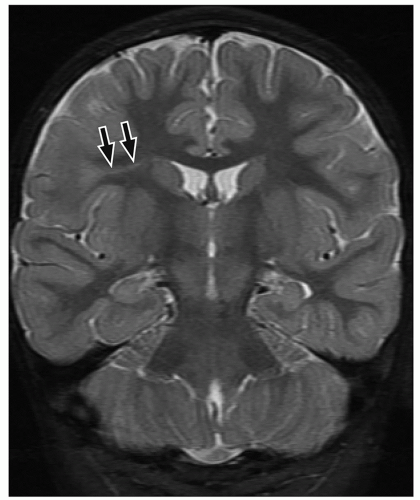 FIGURE 2.13 Focal cortical dysplasia type IIb. Coronal T2-weighted MR image shows a band of hyperintensity (arrows) extending from the gray and white matter interface to the surface of the ventricles in the right frontal lobe. |
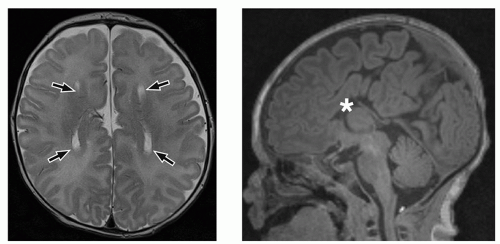 FIGURE 2.14 Agenesis of the corpus callosum. Axial T2-weighted (on the left) and sagittal MPRAGE (magnetization-prepared rapid acquisition gradient-echo) (on the right) MR images show the parallel orientation of the lateral ventricles (arrows) in the axial plane and absence of the corpus callosum in its expected position (asterisk) in the sagittal plane. |
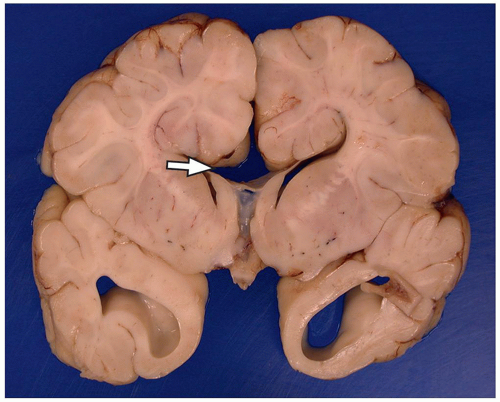 FIGURE 2.15 Agenesis of the corpus callosum. Coronal section at the level of the basal ganglia shows an absent corpus callosum (arrow) in an 11-month-old boy with Vici syndrome, which is also called immunodeficiency with cleft lip/palate, cataract, hypopigmentation, and absent corpus callosum. |
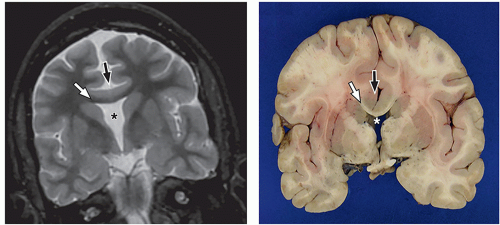 FIGURE 2.16 Lobar holoprosencephaly in an infant girl. Coronal T2-weighted MR image (on the left) and cut surface of the brain (on the right) show fusion of the cortex across the midline (black arrows), absent fornices and septum pellucidum (asterisks), and a thin corpus callosum (white arrows). |
cerebellum (i.e., cerebellar hypoplasia). In the severe variant, both hemispheres and vermis are almost completely absent. The brainstem, particularly the pons, is hypoplastic.
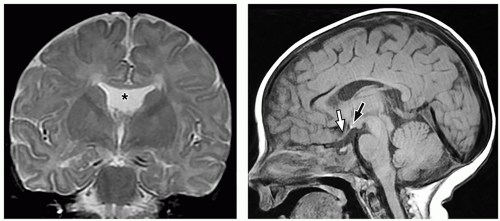 FIGURE 2.17 Septooptic dysplasia. Axial T2-weighted (on the left) and sagittal T1-weighted (on the right) MR images show absent septum pellucidum (asterisk), ectopic posterior pituitary bright spot (black arrow), and hypoplastic optic chiasm (white arrow). |
to the folia and is usually associated with hypoplasia of the pons. In contrast, cerebellar atrophy refers to a small cerebellum with prominent cerebellar fissures or evidence of progressive volume loss shown on serial imaging.
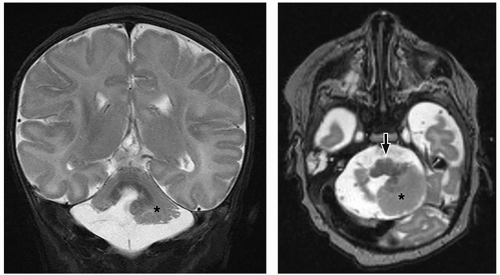 FIGURE 2.18 Cerebellar hypoplasia. Coronal (on the left) and axial (on the right) T2-weighted MR images demonstrate marked right cerebellar hypoplasia, with milder hypoplasia of the left cerebellar hemisphere (asterisks) and small inferior vermis with a small flattened pons (arrow). Note the abnormal cortical outline in the hypoplastic cerebellar hemispheres. |
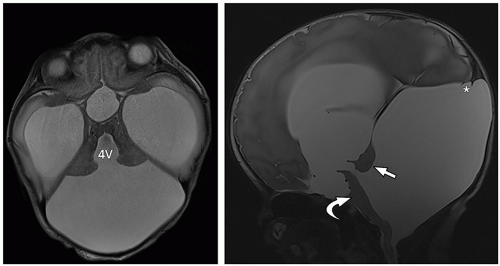 FIGURE 2.19 Dandy-Walker malformation in a 2-year-old infant boy with severe hydrocephalus. Axial (on the left) and sagittal (on the right) T2-weighted MR images show a markedly enlarged CSF-filled posterior fossa, which communicates with an enlarged fourth ventricle (4V), upward rotated hypoplastic vermis (white straight arrow), high tentorium, hypoplastic brainstem (curved arrow), and elevation of the confluence of the venous sinuses (asterisk). |
thalami, fornices, and tectum may be present. Other midline and forebrain anomalies including absent cavum septum pellucidum, absent olfactory bulbs, and corpus callosum dysgenesis can be also seen.
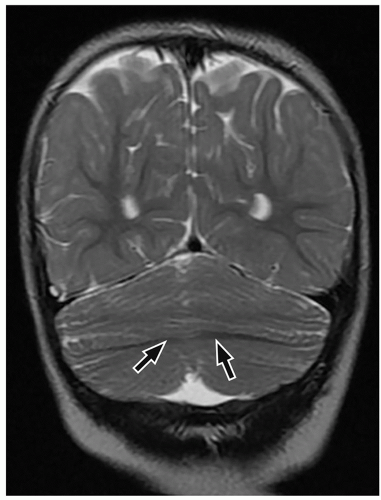 FIGURE 2.20 Rhombencephalosynapsis. Coronal T2-weighted MR image shows apparent fusion of the cerebellar hemispheres with transversely oriented folia and continuity (arrows) of the cerebellar white matter across the midline. |
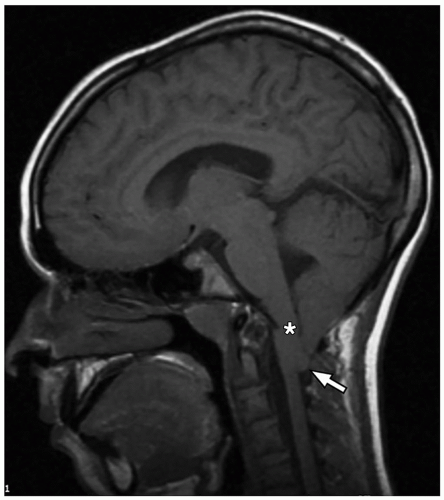 FIGURE 2.21 Chiari I malformation. Sagittal T1-weighted MR image shows pointed cerebellar tonsils (arrow) extending inferiorly to the mid C2 level, with resultant effacement of the CSF spaces at the level of the foramen magnum and associated kinking (asterisk) of the cervicomedullary junction. |
urgent evaluation. Vocal cord abduction, paresis, or paralysis resulting from dysfunction of the vagus nerve causes the inspiratory stridor. Cranial nerve dysfunction is attributed to various causes including caudal traction on the nerve by the herniated medulla/medullary kink, lower brainstem compression, or an abnormally formed dorsal motor nucleus within the brainstem.
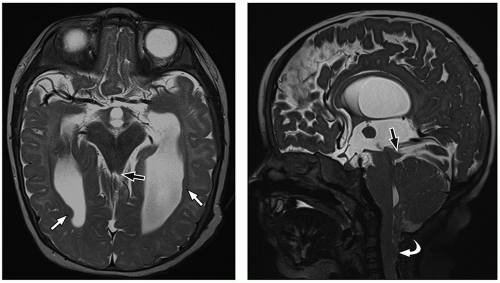 FIGURE 2.22 Chiari II malformation. Axial (on the left) and sagittal (on the right) T2-weighted MR images show a small posterior fossa, herniation of the cerebellar tonsils (curved arrow) to the C4 level with mild kinking of the cervicomedullary junction, tectal beaking (black arrows), and associated subependymal gray matter heterotopia (white arrows). |
containing only CSF), and atretic cephaloceles (containing dura, fibrous tissue, and degenerated brain tissue).
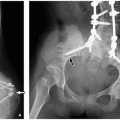
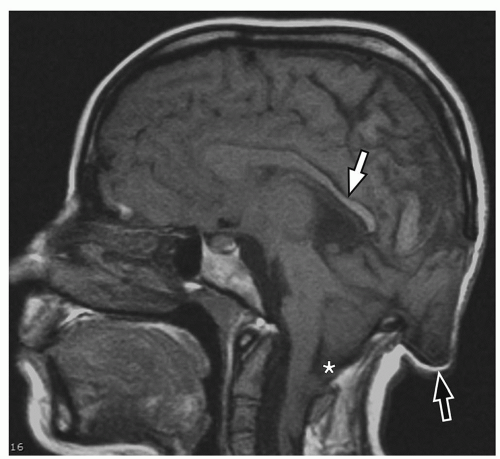 FIGURE 2.23 Chiari III malformation. Sagittal T1-weighted MR image demonstrates an occipital encephalocele (black arrow), which spontaneously decompressed partially after spinal fusion, low lying cerebellar tonsils (asterisk), and thinned posterior half of corpus callosum (white arrow). |
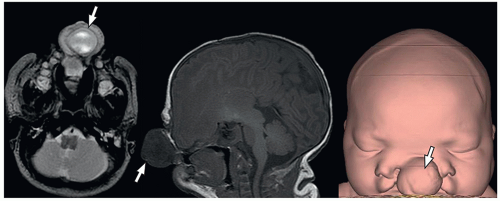 FIGURE 2.24 Frontoethmoidal encephalocele. Axial T2-weighted MR (on the left), sagittal T1-weighted MR (in the center), and 3D reconstructed CT (on the right) images show a large frontoethmoidal cephalocele (arrows) containing dysplastic brain tissue, associated agenesis of corpus callosum, marked hypertelorism, abnormal nose, and cleft palate and lip. |
Arachnoid cysts and neuroepithelial cysts are discussed in the following section. The information regarding leptomeningeal cysts is included in Chapter 1 of this book.
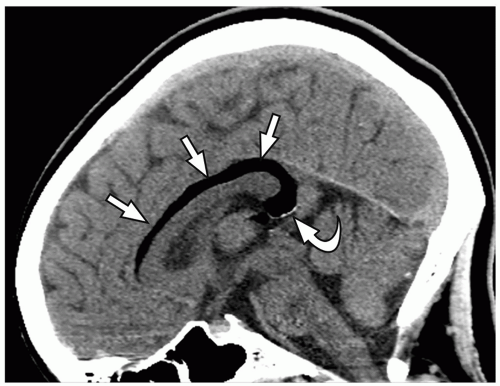 FIGURE 2.25 Corpus callosum lipoma. Sagittal reformatted CT image shows a low attenuation (-50 HU) lesion (straight arrows) in the midline in the pericallosal cistern with a small amount of calcification (curved arrow) along its posterior aspect near the splenium, consistent with a callosal lipoma. |
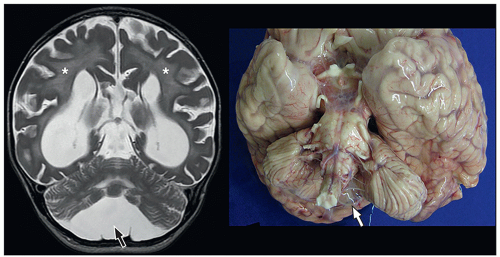 FIGURE 2.26 Arachnoid cyst in a 3-year-old boy with multiple sulfatase deficiency. Coronal T2-weighted MR image (on the left) and postmortem brain specimen (on the right) show a CSF signal intensity thin-walled translucent cyst (black and white arrows) overlying the left inferior surface of the cerebellum, with associated cerebellar asymmetry. Note the abnormal diffuse white matter signal intensity (asterisks) and volume loss in the supratentorial brain on MR image, which is a manifestation of the underlying metabolic disease. |
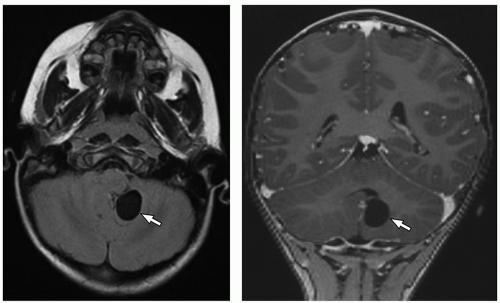 FIGURE 2.27 Neuroepithelial cyst incidentally found in a 10-year-old boy. Axial FLAIR MR image (on the left) and coronal postcontrast T1-weighted SPACE (Sampling Perfection with Application optimized Contrasts using different flip angle Evolution) MR image (on the right) show a cystic CSF signal intensity lesion (arrows) in the cerebellum closely related to the margins of the fourth ventricle. |
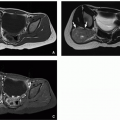
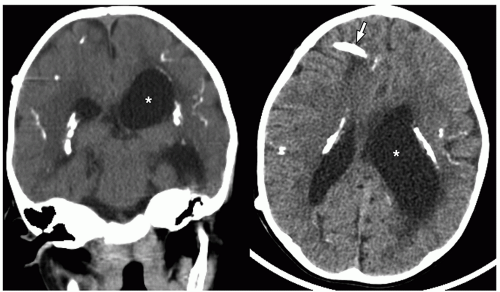 FIGURE 2.28 Congenital toxoplasmosis in an 11-month-old boy. Coronal reformatted unenhanced CT image (on the left) shows multiple foci of intraparenchymal and subependymal calcification. The child also had hydrocephalus (asterisk), which had been shunted by this time. Axial unenhanced CT image (on the right) shows a right frontal shunt catheter (arrow) and hydrocephalus (asterisk). |
TABLE 2.3 Other Congenital TORCH Infections | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
prevent fetal transmission of the CMV virus, but do play a role in reducing severity of disease. Most infants have silent infections following recurrent rather than primary maternal infection. CMV is considered the leading infectious cause of sensorineural hearing loss in the post-rubella vaccination era. Sensorineural hearing loss occurs in around 10% of infected neonates.30 Additional clinical manifestations in affected infants include microcephaly, chorioretinitis, and seizures.
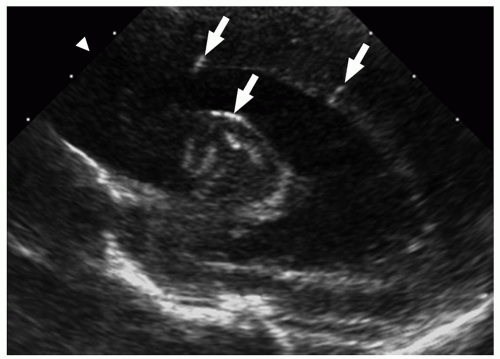 FIGURE 2.29 Congenital cytomegalovirus infection in a 2-day-old boy. Parasagittal ultrasound image shows areas of increased echogenicity consistent with periventricular calcifications (arrows). |
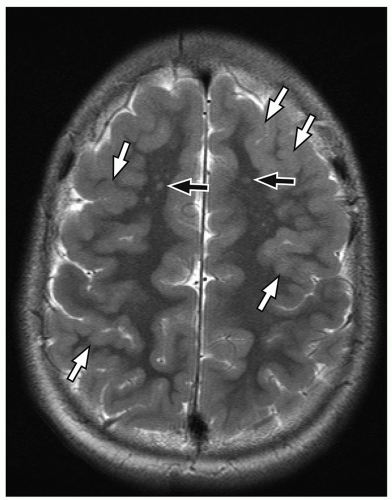 FIGURE 2.30 Congenital cytomegalovirus (CMV) infection. Axial T2-weighted MR image shows extensive frontal polymicrogyria (white arrows) and subcortical heterotopia (black arrows) as a manifestation of congenital CMV. |
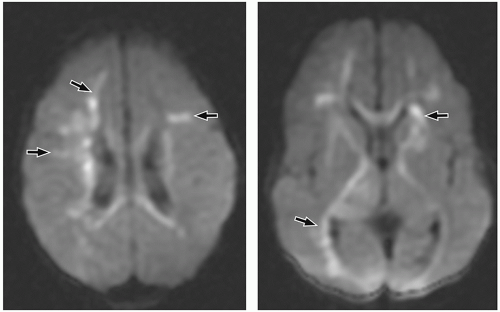 FIGURE 2.31 Neonatal herpes encephalitis in a 12-day-old girl. Axial diffusion-weighted MR images show increased signal (indicating decreased diffusion [arrows]) in the periventricular and deep frontal white matter (on the left), corona radiata, basal ganglia, and internal capsule (on the right). |
puncture, it is important to recognize that normal CT findings may not be sufficient to indicate normal intracranial pressure in pediatric patients with bacterial meningitis. Review of the literature indicates that herniation is unlikely in children with bacterial meningitis unless they have focal neurologic findings or coma.32 In addition, the results of an imaging study do not exclude or prove the presence of acute meningitis. Diagnosis should therefore be made based on clinical history, examination findings, and results of laboratory tests.
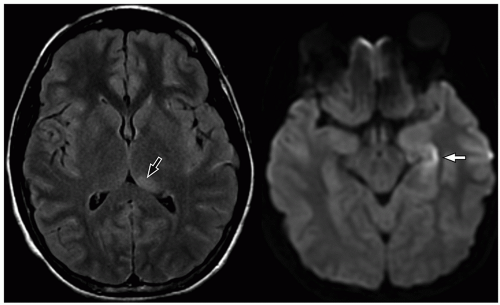 FIGURE 2.32 Herpes simplex virus encephalitis in a 9-year-old girl. Axial FLAIR MR image (on the left) and axial diffusion-weighted MR image (on the right) demonstrate signal abnormality in the left thalamus (black arrow) and diffusion restriction in the left hippocampus (white arrow). The distribution of findings is most suggestive of herpes encephalitis, which was proven by polymerase chain reaction analysis of CSF. |
more commonly than adults. Organisms from these foci are released into the subarachnoid space causing meningitis. Meningitis is typically most severe in the basal cisterns and leads to secondary complications, including multiple cranial nerve palsies, vasculitis of the lenticulostriate and thalamoperforating vessels resulting in secondary infarction of the basal ganglia, and hydrocephalus secondary to blockage of the fourth ventricular outlet foramina. It is important to note that hydrocephalus in patients with tuberculous meningitis may need to be treated surgically. This can be determined with an emergent CT at presentation, followed by MRI later.34 Imaging using diffusion-weighted MRI is important to document the presence of infarcts, which correlates with poor outcome. In particular, bilateral basal ganglia infarcts have a poor prognosis. Border zone necrosis seen in these patients needs to be differentiated from necrosis in older children with herpes, which is most often seen at the insular cortex.35
 FIGURE 2.33 Recurrent meningitis in a 3-year-old girl. Axial CT image (on the left) shows posterior fossa dermoid (white arrow). A tract (black arrow) passing through occipital bone on to the overlying scalp is also seen (in the middle). This was not noticed at the time of the CT study, and at a subsequent presentation, axial postcontrast T1-weighted MR image (on the right) shows infected material within the dermoid (curved arrow) and an adjacent abscess (A) in the cerebellar hemispheres. |
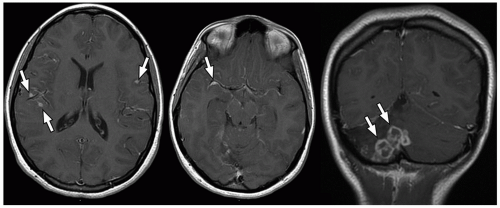 FIGURE 2.34 Tuberculous meningitis and meningoencephalitis in a 17-year-old girl. Axial and coronal postcontrast T1-weighted MR images show abnormal linear and nodular meningeal enhancement (arrows) in the bilateral temporal lobes and basal cisterns (on the left and in the middle), and multiple foci of ring enhancement (arrows) in the right cerebellar hemisphere with surrounding encephalomalacia (on the right). |
perimesencephalic cisterns, suprasellar cisterns, and sulci over the convexities are commonly affected.
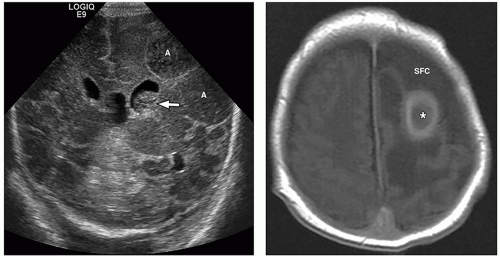 FIGURE 2.35 Enterobacter cerebral abscess. Coronal cranial ultrasound image (on the left) shows heterogeneous left frontoparietal intraparenchymal lesions (A) with surrounding increased echogenicity and sulcal effacement, consistent with cerebral abscess. Also noted is an echogenic lesion (arrow) in the left lateral ventricle representing intraventricular pus. Subsequently obtained axial postcontrast MR image (on the right) shows a rim-enhancing lesion (asterisk) with adjacent subdural fluid collection (SFC) overlying the left frontal lobe. |
demonstrate enhancement of the walls of the lesion following gadolinium-based contrast administration (Fig. 2.35). Diffusion-weighted MR imaging is helpful to differentiate between an abscess and a necrotic tumor. Therefore, it must be performed in all cases of suspected CNS infection because almost all pyogenic abscesses demonstrate decreased apparent diffusion coefficient (ADC) values, indicating decreased water diffusion compared with nonpyogenic lesions.
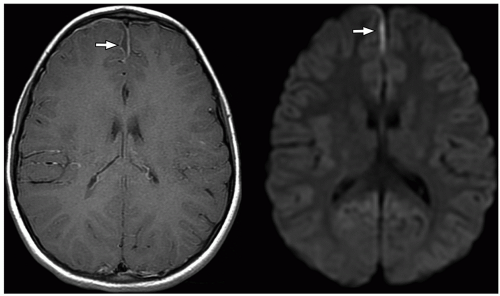 FIGURE 2.36 Subdural empyema as a complication of sinusitis in a 7-year-old girl. Axial postcontrast T1-weighted MR image (on the left) and axial diffusion-weighted MR image (on the right) show a rim-enhancing collection with restricted diffusion, consistent with a right frontal parafalcine subdural empyema (arrows). |
detect early fibrosis within the inner ear structures and labyrinthitis ossificans33 (Fig. 2.38).
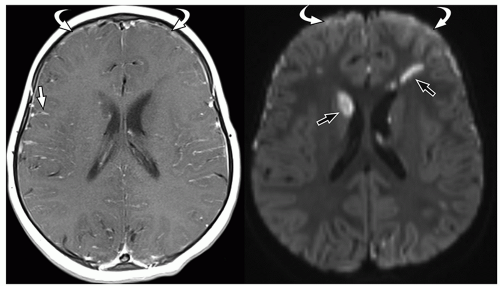 FIGURE 2.37 Pneumococcal meningitis in a 22-month-old girl. Axial postcontrast T1-weighted MR image (on the left) shows leptomeningeal enhancement (straight arrow) and small frontal enhancing subdural collections (curved arrow). Axial diffusion-weighted MR image (on the right) shows multiple foci of decreased diffusion (straight arrows) in the basal ganglia and frontal white matter, without corresponding enhancement on postcontrast MR images, indicating that these are infarcts secondary to meningitis. Note decreased diffusion in the bifrontal subdural empyemas (curved arrows). |
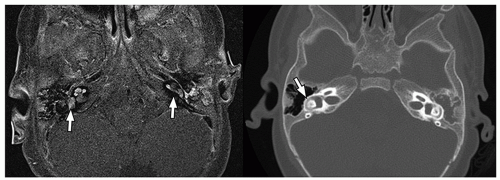 FIGURE 2.38 Labyrinthine enhancement in an 8-month-old boy with pneumococcal meningitis. Axial postcontrast T1-weighted fat-saturated MR image obtained at initial presentation (on the left) shows enhancement (arrows) within the inner ear structures bilaterally. Subsequently obtained CT 2 months later, just before cochlear implantation (on the right), shows subtle increased density (arrow) in the right lateral semicircular canal, consistent with early labyrinthitis ossificans. |
tumors in childhood is ˜4.5 cases per 100,000 person-years.42 In young children <3 years of age, supratentorial tumors are more common than infratentorial tumors.42 In children between 4 years and 10 years of age, infratentorial tumors occur more frequently. Supra- and infratentorial tumors occur equally after 10 years old age.42
TABLE 2.4 Clinical Manifestations and Central Nervous System Imaging Features of Fungal Infections | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 2.5 Common Anatomic Locations of Pediatric Brain Tumors | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
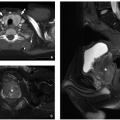
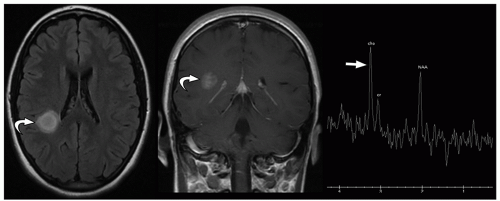 FIGURE 2.39 Hemispheric pilocytic astrocytoma. Axial FLAIR MR image (on the left), postcontrast coronal T1-weighted MR image (in the middle), and single voxel MR spectroscopy image (on the right) show a rounded enhancing T2 hyperintense lesion (curved arrows) in the right parietal white matter, with elevated choline peak on MR spectroscopy (straight arrow). |
be seen (Fig. 2.42). Intratumoral bleeding is also common because of the abnormal, rich vasculature that characterizes these tumors. Histologically, glioblastoma multiforme consists of poorly differentiated glial cells, often with pronounced variation in nuclear size and shape (anaplasia or pleomorphism).
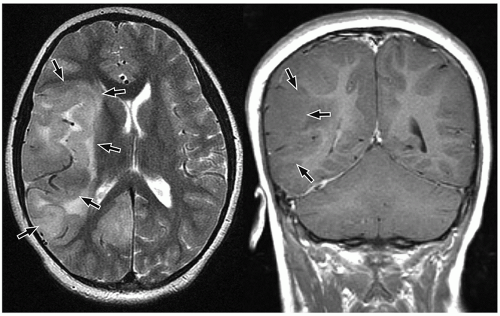 FIGURE 2.40 Gliomatosis cerebri in a 7-year-old girl who presented with confusion and left-sided weakness. Axial T2-weighted MR image (on the left) and coronal postcontrast T1-weighted MR image (on the right) show a large mass (arrows) characterized by abnormal T2 signal occupying large portions of the right cerebral hemisphere with mass effect. |
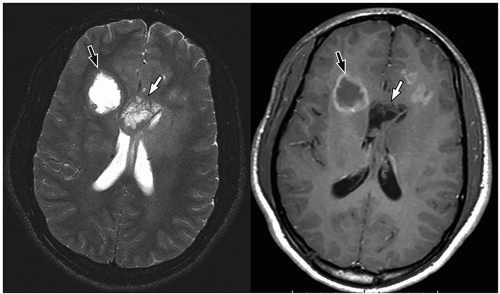 FIGURE 2.41 Glioblastoma multiforme in a 16-year-old boy. Axial T2-weighted MR image (on the left) and axial postcontrast T1-weighted MR image (on the right) show a large frontal lobe mass (black arrows) with a T2 hyperintense central necrotic core and enhancing margins. In addition, there is an extensive surrounding T2 hyperintense lesion extending across the midline (white arrows), consistent with the “butterfly glioma” characteristic of glioblastoma multiforme. |
slow-growing peripherally located lesions, remodeling of the inner table of the skull is a common finding.
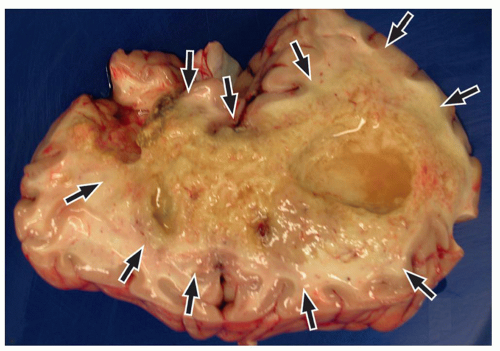 FIGURE 2.42 Glioblastoma multiforme in a 16-month-old boy who was diagnosed 3 months antemortem. The cut surface shows a large mass (arrows) with multiple areas of yellow-hued necrosis and an ill-defined border. |
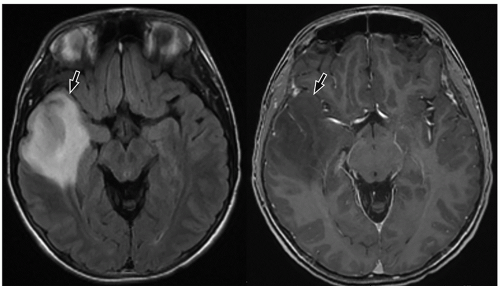 FIGURE 2.43 Oligodendroglioma in a 15-year-old boy. Axial FLAIR MR image (on the left) and axial T1-weighted SPGR (spoiled gradient recalled) MR image (on the right) demonstrate a FLAIR hyperintense lesion in the right temporal lobe that does not enhance following contrast administration (arrows). Histology of the resected specimen revealed an oligodendroglioma. |
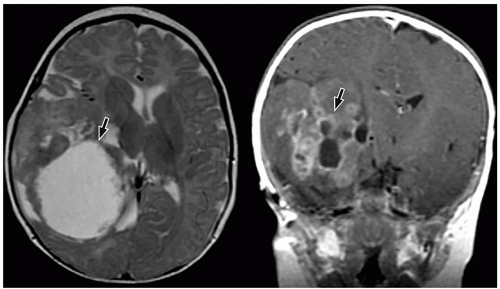 FIGURE 2.44 Supratentorial ependymoma in a 5-month-old girl. Axial T2-weighted MR image (on the left) and coronal postcontrast T1-weighted MR image (on the right) show a large heterogeneous mass (arrows) containing a large cystic component and heterogeneous enhancement in the right cerebral hemisphere, with vasogenic edema and marked mass effect on surrounding brain. Histology of the resected specimen was consistent with an anaplastic ependymoma. |
in young adults. Both tumors arise in the cerebral cortex, most commonly in the temporal lobe. Presenting symptoms depend upon the size and location of tumors and usually include seizures. In particular, complex partial seizures are commonly associated with temporal lobe tumors.
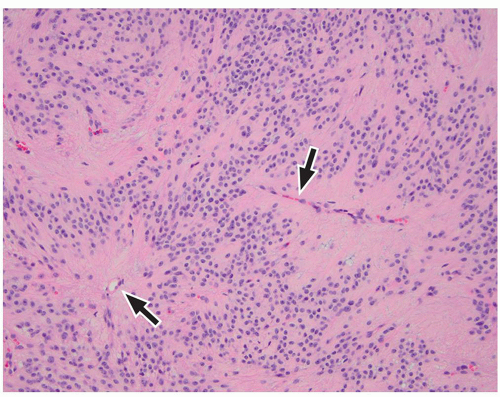 FIGURE 2.45 Ependymoma in a 6-year-old girl. Histology from a mass resected from the frontal lobe shows monotonous round cells in a fibrillary background. Tumor cell nuclei are often arranged around hypocellular fibrillary areas with central vessels (arrows), forming “perivascular pseudorosettes” (hematoxylin and eosin, original magnification, 200×). |
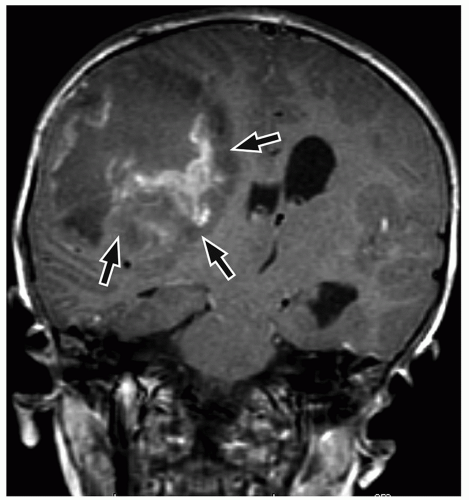 FIGURE 2.46 Central nervous system (CNS) embryonal tumor (diagnosed as “CNS PNET” before WHO nomenclature was changed) in an 8-year-old boy. Coronal postcontrast T1-weighted MR image demonstrates a large, partially necrotic, enhancing mass (arrows) in the right cerebral hemisphere causing marked mass effect and midline shift. |
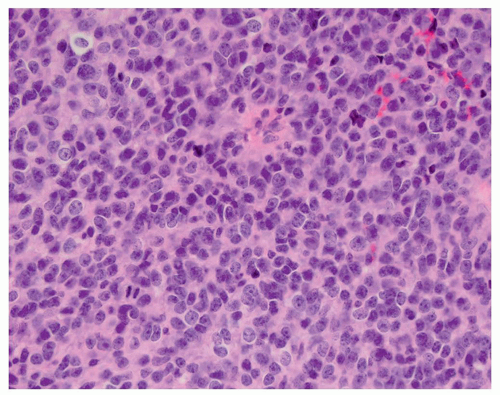 FIGURE 2.47 Embryonal tumor from the temporal lobe of a 3-year-old girl. Poorly differentiated densely packed cells with high nuclear-to-cytoplasmic ratio are present (hematoxylin and eosin, original magnification, 400×). Genetic findings may permit specific classification, such as “embryonal tumor with multilayered rosettes, C19MC-altered”; when not further classifiable, the designation “CNS embryonal tumor, not otherwise specified” is appropriate, as the old term “CNS PNET” is now defunct. |
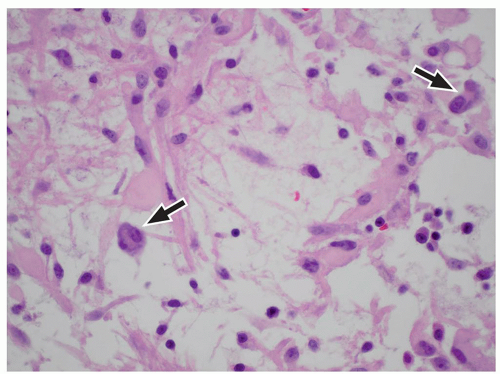 FIGURE 2.48 Ganglioglioma in a 14-year-old boy. This mass resected from the parietal lobe is moderately cellular with a spindled glial component and admixed dysplastic, occasionally multinucleate (arrows) ganglion cells (hematoxylin and eosin, original magnification, 400×). |
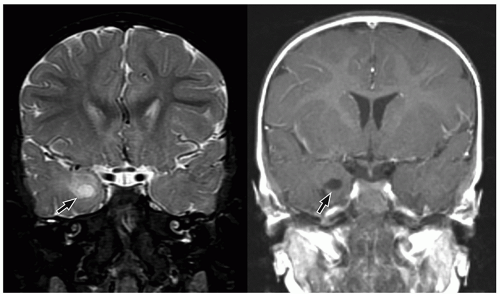 FIGURE 2.49 Ganglioglioma in an 11-month-old girl who presented with seizures. Coronal T2-weighted MR image (on the left) and coronal postcontrast T1-weighted MR image (on the right) show a poorly-defined, nonenhancing, heterogeneous mass (arrows) that is hyperintense to gray matter expanding the right temporal lobe, suggestive of a ganglioglioma. |
of enhancement may suggest the diagnosis of these tumors, although the appearance is still nonspecific.
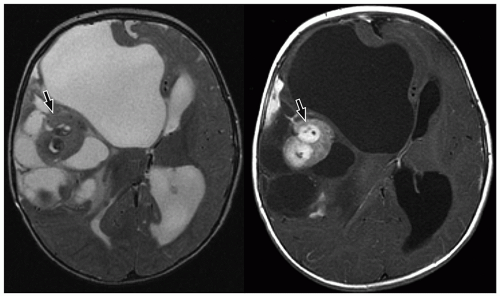 FIGURE 2.50 Desmoplastic infantile ganglioglioma in an asymptomatic 9-month-old boy who presented with increasing head circumference. Axial T2-weighted MR (on the left) and axial postcontrast T1-weighted MR image (on the right) show a large mixed solid and cystic lesion (arrows) in the right cerebral hemisphere, with enhancement of some of the solid components. Mass effect from the tumor and leftward midline shift are also present. |
CNS tumors in childhood.42 DNETs are located most commonly in the temporal and parietal lobes. Most lesions arise from the cortical gray matter, and associated cortical dysplasia has been reported in more than 80% cases.51,52
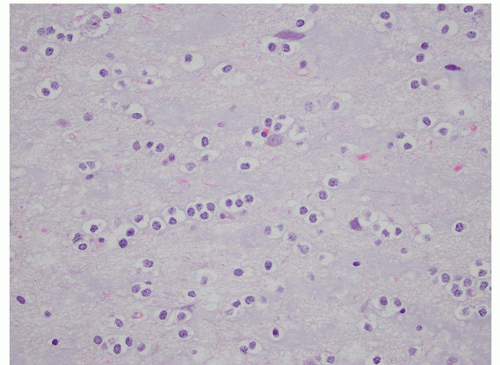 FIGURE 2.51 Dysembryoplastic neuroepithelial tumor in a 15-year-old boy. Tumor resected from the temporal lobe shows round oligodendrocyte-like cells with occasional interspersed neurons, present in a pale blue myxoid background (hematoxylin and eosin, original magnification, 400×). |
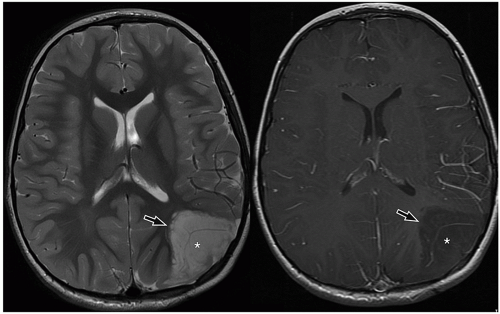 FIGURE 2.52 Dysembryoplastic neuroepithelial tumor in a 10-year-old boy who presented with seizures. Axial T2-weighted MR image (on the left) and axial postcontrast T1-weighted MR image (on the right) show a large cortically based lesion (asterisks) with cyst-like areas (arrows) along the medial aspect, which does not enhance on the postcontrast MR image. |
bilateral optic nerve tumors are virtually pathognomonic of this diagnosis. Optic pathway gliomas may involve the optic nerves, optic chiasm, optic tract, lateral geniculate bodies, and/or optic radiations. Optic gliomas occur with increasing frequency in patients with NF1 (20% to 50%).56 On the other hand, up to 24% of patients with NF1 have optic pathway gliomas.57 Tumors in children with NF1 are reportedly less aggressive than those in children without NF1 and tend to be bilateral.
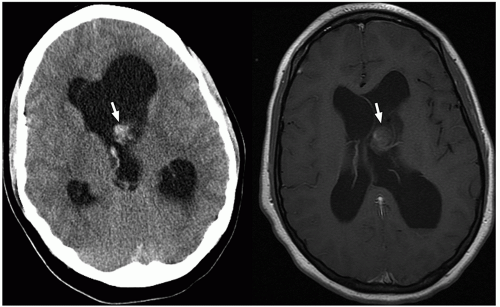 FIGURE 2.53 Neurocytoma in a 14-year-old girl. Axial noncontrast CT image (on the right) and axial postcontrast T1-weighted MR image (on the left) show a hemorrhagic, partially enhancing tumor (arrows) arising from the left head of caudate with intraventricular extension. |
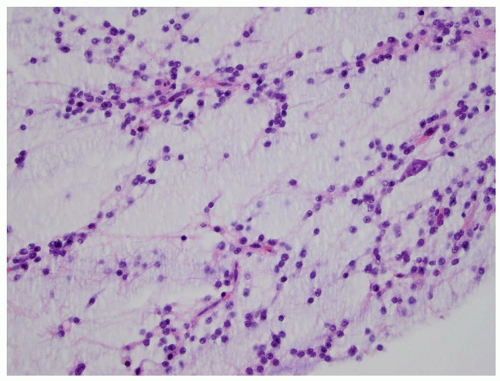 FIGURE 2.54 Neurocytoma. This intraventricular mass from the same patient (Figure 2.53) shows round uniform cells interspersed with “nucleus-free” zones of fibrillary neuropil (hematoxylin and eosin, original magnification, 400×). |
The adamantinomatous type is more common in children, whereas the squamous-papillary variant tends to occur in adults.59,60 Although they are benign, they can invade surrounding structures in the sellar and parasellar regions, eliciting a gliotic response and making resection challenging.
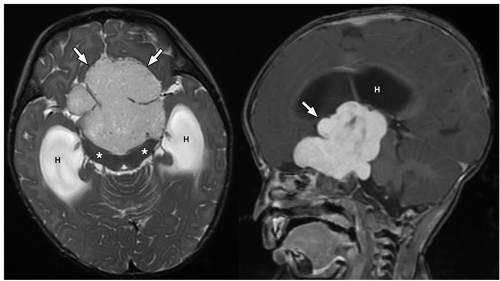 FIGURE 2.55 Optic pathway glioma in a 6-month-old girl who presented with increased irritability and was diagnosed with diencephalic syndrome. Axial T2-weighted MR image (on the left) and postcontrast sagittal 3D SPGR (spoiled gradient recalled) MR image (on the right) show an avidly enhancing suprasellar and sellar intermediate T2 signal intensity lesion (arrows) extending to the prepontine region and splaying the cerebral peduncles (asterisks) consistent with a large optic pathway glioma with secondary obstructive hydrocephalus (H). |
ng/mL.65 Functioning microadenomas are usually treated with medical therapy, although some surgeons prefer upfront operative management in pediatric patients. Microscopically, pituitary adenomas may be classified based on their architectural pattern and hormonal content as assessed by immunohistochemical staining (Fig. 2.59).
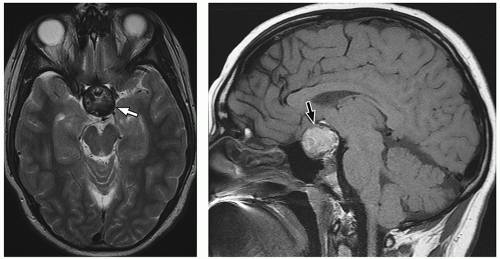 FIGURE 2.56 Craniopharyngioma in a 17-year-old boy who presented with hypopituitarism. Axial T2-weighted MR image (on the left) and sagittal postcontrast T1-weighted MR image (on the right) shows a large, partially calcified, heterogeneous, suprasellar mass (arrows) with mass effect upon the optic chiasm and involvement of the pituitary gland. |
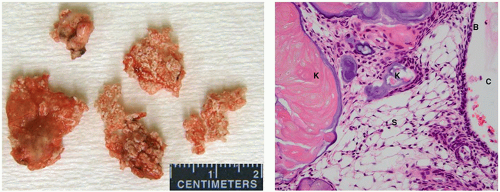 FIGURE 2.57 Craniopharyngioma in a 3-year-old girl. This adamantinomatous variant of a craniopharyngioma was a large cystic hypothalamus-centered mass. Gross examination (on the left) shows spongy and somewhat chalky pale tan tissue with occasional cysts containing dark, greasy fluid. Microscopically (on the right), squamous cells with basal palisading (B) and stellate reticulum (S) alternate with calcifying keratinous debris (K) and cystic spaces (C) (hematoxylin and eosin, original magnification, 400×). |
MR images. Intracystic nodules have also been described, but may be difficult to visualize because of similar signal intensity of the cyst fluid and the nodule. Although imaging findings may be helpful for differentiating these lesions from other sellar or suprasellar lesions, radiologic findings can be nonspecific, and features can overlap with cystic craniopharyngiomas or cystic pituitary adenomas. Follow-up imaging is required in many cases, and occasionally, cyst aspiration may be required to establish a diagnosis.
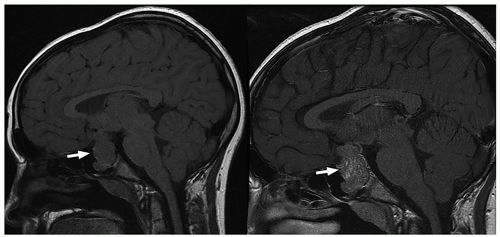 FIGURE 2.58 Pituitary macroadenoma in a 16-year-old boy who presented with growth hormone deficiency. Sagittal precontrast T1-weighted MR image (on the left) and sagittal postcontrast T1-weighted MR image (on the right) through the sella show a large, heterogeneous, partially-enhancing macrolobulated lesion (arrows) expanding the sella and extending superiorly into the suprasellar cistern and inferiorly into the sphenoid sinus. |
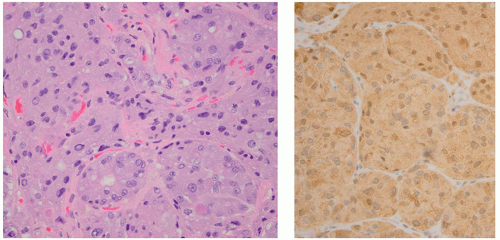 FIGURE 2.59 Pituitary adenoma in a 13-year-old boy who presented with Cushing disease. Resected mass demonstrates characteristic nests of cells showing abundant variably granular cytoplasm (left, hematoxylin and eosin, original magnification, 400×). Immunohistochemical staining shows brown cytoplasmic reactivity for adrenal corticotropic hormone (right, ACTH immunostain, original magnification, 400×). |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


