Although the exact incidence is not known, the majority of bone tumors in children are benign.
13 It is common for benign bony lesions to be found incidentally on radiographs obtained for evaluation of traumatic injury or joint pain. For benign bone lesions, such as benign fibrous cortical defects (FCDs), or nonossifying fibromas (NOFs), radiography alone is sufficient for diagnosis, and there is no need for further imaging studies or biopsy.
Benign bone lesions are typically well-demarcated, having a narrow transition zone with sclerotic rim and no cortical bone breakdown or soft tissue extension. In some cases, the lesions can have an aggressive radiographic appearance that may mimic malignant bone tumor or osteomyelitis, requiring further evaluation.
14 CT is superior to radiography or MRI in visualizing matrix calcification or ossification and associated fracture. MRI is excellent for showing the cross-sectional intraosseous and extraosseous extent of bone tumors, internal characteristics such as necrosis, hemorrhage, or fluid-fluid levels, adjacent marrow edema, and enhancement pattern.
Osteochondroma
Osteochondroma, also known as an osteocartilaginous exostosis, is a cartilage-capped bony excrescence that arises from the external surface of the bone and contains a medullary cavity that is continuous with that of the parent bone
15 (
Fig. 23.1). It is the most common benign bone tumor, accounting for 20% to 50% of benign bone tumors and affecting 1% of the general population.
1 In most cases, osteochondromas present as a solitary bone lesion (86% of cases) and occur in the first three decades of life with slight male predilection (male: female 1.6 to 3.4:1).
1 They most commonly arise from the metaphyses of the long bones, particularly the distal femur, proximal tibia, and proximal humerus. Less commonly involved locations are the radius, fibula, and flat bones including the ileum, scapula, and ribs. Involvement of small bones of the hand or the foot is seen in 10% of osteochondromas.
16Multiple osteochondromas account for 14% of cases and occur in the setting of hereditary multiple exostoses (HME). In ˜80% of solitary osteochondromas, homozygous mutations in the
EXT1 gene can be found in the cartilaginous cap. In HME, also known as familial osteochondromatosis or diaphyseal aclasis, germline mutations in the
EXT1 or
EXT2 gene are commonly identified.
17 HME most commonly
affects the distal and proximal bones of the lower extremities, and involvement is typically bilateral and symmetrical. Nearly 2% of patients with HME eventually develop a chondrosarcoma.
17Osteochondroma is the most common tumor in irradiated skeletons; 6% to 12% of patients who underwent previous radiation at a young age develop osteochondromas, and latent periods are variable with a range of 3 to 16 years.
18,
19 Imaging findings and histologic features are the same as those of primary osteochondroma.
In the immature skeleton, osteochondromas continue to increase in size by endochondral ossification occurring at the base of the cartilage cap, which is equivalent to the epiphyseal physis, and growth ceases after skeletal maturation (physeal closure). Spontaneous regression of osteochondromas has been reported after skeletal maturation. In many cases, osteochondromas are asymptomatic and found incidentally, but affected pediatric patients can present with pain and other symptoms. Complications of osteochondromas include fracture, bursa formation, bursitis, vascular injury, and neurologic compromise. Complications and osseous deformities are more common in HME.
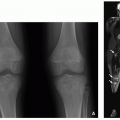
The imaging findings of osteochondromas are characteristic and include pedunculated or sessile lesions. Pedunculated osteochondromas have a long and slender appearance with a bony excrescence arising from the surface of the bone (usually long bone metaphyses). Pedunculated osteochondromas grow in the direction of tendon pulling (
Fig. 23.2). Sessile osteochondromas have a broad-based and flat appearance. Typically, the medullary portion of the osteochondroma merges into the bone marrow of the parent bone, and the bony cortex and periosteum continuously outline both parent bone and osteochondroma. In contrast to osteochondroma, juxtacortical osteosarcoma
has interposed periosteum and cortex between tumor and parent bone.
Imaging findings of individual osteochondromas in HME and solitary osteochondromas are the same. Besides multiple osteochondromas, HME is characterized by abnormal modeling of developing bone, including metaphyseal widening, growth disturbance, and deformities of the joints. Radiographic evaluation is required to delineate the dramatic bone changes.
20 The affected bone is abnormally wide due to failure of normal tubulation. The cartilage cap is not visualized on radiographs except when irregular zones of calcification occur in the cartilage cap. Small stippled calcifications are compatible with a benign growth of the cartilage cap.
21 Osteochondromas arising from the innominate bone tend to be especially large and may exert mass effect, displacing adjacent structures.
Both CT and MRI can show the continuity of the medullary bone between the parent bone and the lesion. MRI is superior to CT in demonstrating the cartilaginous cap, which has a curvilinear area of fluid-like signal intensity (low signal intensity on T1-weighted MR images and high signal intensity on T2-weighted MR images), and can allow accurate measurement of the thickness of the cartilage cap (
Fig. 23.2). In children and adolescents, the cartilage cap can be as thick as 3 cm.
22 In adults, the cartilage cap is usually <1 cm in thickness or can be entirely absent. In skeletally mature patients, a cartilage cap presenting with more than 1.5 cm in thickness raises the possibility of underlying malignant change.
23Malignant transformation to chondrosarcoma can occur in the cartilage cap and is reported in <1% of solitary osteochondromas and in nearly 2% of HME (
Fig. 23.3).
24 Imaging features suggesting malignant transformation include growth of the lesion in a skeletally mature patient or after physeal closure, irregular or indistinct lesion surface, focal areas of radiolucency in the base of the lesion, erosion or destruction of the adjacent bone, and new soft tissue formation with extensive or irregular calcification (
Fig. 23.3).
24 Malignant transformation of osteochondroma to chondrosarcoma before the age of 20 years is unusual. Malignant transformation of osteochondroma to osteosarcoma, which usually occurs in the osteochondroma stalk, is even rarer.
Small asymptomatic osteochondromas do not require treatment. Surgical resection is reserved for large solitary lesions or lesions with complications. In pediatric patients with HME, treatment is complex to correct deformities as well as to resect lesions, and often multiple surgical procedures are required.
Osteochondroma Variants
Dysplasia epiphysealis hemimelica (DEH), also known as Trevor disease, is a rare developmental disorder characterized by osteochondromas arising from the epiphysis. DEH is nonhereditary and more commonly seen in boys. The lower extremity is usually affected, and tarsal or carpal bones are also involved in some cases.
25 DEH is caused by asymmetric overgrowth of the medial or lateral aspect of the epiphyseal cartilage or epiphyseal equivalent. Three different types are observed: localized (single epiphyseal involvement), classical (involvement of more than one epiphysis of the same limb), and generalized (involvement of the entire limb).
26Radiographs usually demonstrate the characteristic appearance of asymmetric epiphyseal cartilage overgrowth with multiple stippled, irregular, and dense calcifications in the epiphysis.
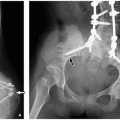
27 The lesion is more likely to be medial than lateral in a given bone part, and CT can clearly show the continuity of the mass with the mother bone (
Fig. 23.4). MRI is helpful for defining epiphyseal overgrowth, which is mainly cartilaginous before ossification. On MRI, the osteochondroma appears incorporated into the epiphysis, and the lesion has the same signal intensity as normal cartilage and may contain internal low signal spots from calcified foci.
Subungual exostosis is a neoplastic overgrowth of cartilage and trabecular bone in the nail bed of the phalanx. Bizarre parosteal osteochondromatous proliferation (BPOP), also known as Nora lesion, is a benign proliferation of bone and cartilage that arises from the bone surface in the hand or foot, or less commonly the long bones. BPOP presents with a nodular mineralized mass arising from the hand and foot bones (
Fig. 23.5).
24
Simple Bone Cyst
A simple bone cyst (SBC), also known as a unicameral cyst, solitary bone cyst, or juvenile bone cyst, is an intramedullary membrane-lined, fluid-filled cavity in the bone. In most cases, SBC remains asymptomatic and is discovered incidentally. However, SBC may present with pain and swelling when there is a pathologic fracture. SBC is most commonly found in the proximal humerus (over two-thirds of cases), followed by the proximal femur. SBC usually involves the metaphysis and is centered in the medullary cavity, in contrast to the eccentric location of a typical aneurysmal bone cyst (ABC). As the child grows, SBC appears to gradually migrate away from the physis into the diaphysis.
On radiograph, SBC appears as a well-demarcated radiolucent lesion with or without septations. There is no or little expansion of the bone. A “fallen-fragment” sign, representing a fractured bone fragment dislodged in the dependent portion of the cyst, is pathognomonic for SBC with pathologic fracture (
Fig. 23.6).
28 On MRI, the bone cyst appears as a high signal intensity fluid-filled lesion on T2-weighted MR images and low to iso-signal intensity on T1-weighted MR images. There may be rim or cyst wall enhancement after administration of gadolinium. When complicated by a pathologic fracture, fluid-fluid levels are often demonstrated within a SBC. If the SBC occurs in the calcaneus, mostly in adults, it is typically located near the neck of the calcaneus.
29 Calcaneal pseudocyst and intraosseous lipoma may have a
similar radiographic appearance with well-demarcated radiolucent lesions.
The current treatment of SBC includes steroid injection after cyst aspiration, sclerotherapy, and curettage with cement engrafting. Posttreatment radiography may show a more complex appearance of the no longer “simple” cyst with sclerosis and deformity.
Aneurysmal Bone Cyst
ABC is a benign bone neoplasm composed of blood-filled spaces separated by multiple thin fibrous septa. Lesions with histologic features of ABC can arise de novo (primary), which accounts for 70% of cases, or they may occur adjacent to other benign or malignant bone tumors (secondary) in the remaining 30%.
4 ABC is most common during the first two decades of life with no gender predilection. The most commonly affected sites are the metaphyses of the long tubular bones, the femur, tibia and humerus, and the posterior aspect of the spine. Typical clinical presentations include pain and swelling and neurologic symptoms in cases of spinal involvement.
Radiographs usually show a well-delineated and expansile osteolytic lesion with thin walls and septa creating a “ soap-bubble” appearance. In the long bones, ABC is usually eccentric in location, and the outer cortex is markedly thin (
Fig. 23.7). CT and MRI can reveal fluid-fluid levels due to blood products; this finding is nearly diagnostic of ABC (
Fig. 23.7). It is important to recognize that secondary ABC components with fluid-fluid levels can be found in various other benign bone lesions such as osteoblastoma, chondroblastoma, giant cell tumor (GCT), NOF, cystic fibrous dysplasia, and SBC. In addition, some of malignant bone lesions such as osteosarcoma, either telangiectatic or even conventional type, can also have an ABC component. Fat-suppressed T2-weighted MR images are best at clearly showing fluid-fluid levels. The bone cortex is preserved in most cases of ABC unless it is associated with a pathologic fracture. The radiologic differential diagnosis of ABC includes GCT, low-grade osteosarcoma, and telangiectatic osteosarcoma in the pediatric population.
Solid-variant ABC, also known as giant cell reparative granuloma when it occurs in the head and neck, is a rare type of ABC that is primarily seen in the craniofacial bones and small tubular bones of the hand and foot. It is uncommonly seen in the long tubular bones.
30 This variant has a wide spectrum of imaging features, from similar to ABC at one end to having aggressive features such as permeative bone destruction and periosteal reaction simulating malignancy at the other end. One-third of the solid-variant ABC lesions are not expansile (nonaneurysmal).
31 MRI shows a persistent solid component with heterogeneous high T1 and T2 signal intensity in an
expansile cystic lesion. Occasional perilesional bone marrow edema can be also observed in the solid-variant ABC.
30Grossly and histologically, ABC typically shows blood-filled cysts with intervening septa that contain fibroblasts, multinucleate giant cells, and variable numbers of admixed osteoblasts and inflammatory cells (
Fig. 23.8). Chromosomal rearrangement involving the
USP6 gene at chromosome 17p13 is seen in primary ABC, illustrating a relationship between ABC and nodular fasciitis, another fibroblastic proliferation characterized by
USP6 rearrangement.
31 ABC is treated with surgical removal of the entire lesion with bone graft if it is required. Local recurrences after resection are rare.
32
Osteoid Osteoma
Osteoid osteoma is a benign bone-forming tumor that is characterized by a relatively small-sized lesion with disproportionally severe pain. Osteoid osteomas account for 10% to 12% of all benign bone neoplasms and 2% to 3% of all primary bone tumors.
4 Most cases (over 75%) occur between ages 5 and 24 years with male predilection (3:1).
4 In more than 50% of cases, the femur or tibia is affected, and the proximal femur including the femoral neck is the single most common site.
4 Other locations include the midshaft or metadiaphysis of the long bones and less commonly the tarsal bones, hand phalanges, and the spine. Osteoid osteomas are subclassified into three subtypes based on location: cortical, medullary, or subperiosteal. Of these, cortical osteoid osteomas are the most common, and subperiosteal ones are the least common.
33The clinical presentation of an osteoid osteoma is characteristic. Affected pediatric patients experience pain that worsens at night and dramatically improves with salicylates. Unfortunately, the pain is often referred to the adjacent joint and occasionally to a site distant from the lesion, resulting in misdirecting of radiographic studies and incorrect clinical diagnosis.
34The characteristic imaging findings of osteoid osteomas include a nidus (core) that is usually located in the cortex and contains a variable degree of mineralization, associated adjacent cortical thickening, and reactive sclerosis in a long bone shaft (
Fig. 23.9).
33 The nidus appears as a central radiodense
area that is surrounded by a lucent rim or a radiolucent focus because it is located in the center of an area of reactive sclerosis.
The nidus is round or oval and usually less than 2 cm in size.
33 CT best delineates the nidus; it has a target appearance with a well-defined round or oval area of lucency and a central mineralized area.
35 MRI shows a low to intermediate T1 and variable T2 signal intensity nidus, depending on the mineralization of the central osteoid. The mineralized nidus has a target appearance on T2-weighted MR images; there is a central dark spot (mineralized) surrounded by peripheral high signal (unmineralized area). The nidus may show strong enhancement after administration of gadolinium (
Fig. 23.9). With intravenous contrast administration, dynamic CT or MR perfusion imaging technique enables the nidus to be more conspicuously visualized.
36 MRI can also show associated bone marrow edema and surrounding soft tissue changes. With recent advances in MRI techniques, current MRI would be comparable or even better than CT for evaluating osteoid osteoma and has the advantage of showing additional findings such as marrow edema, reactive soft tissue changes, and abnormalities of adjacent joints without associated ionizing radiation exposure.
36Intra-articular osteoid osteoma is rare, but somewhat challenging in clinical and imaging diagnosis. An osteoid osteoma near or within the joint typically presents with joint effusion and pain mimicking arthritis. In contrast to osteoid osteoma in usual locations, reactive cortical thickening may be minimal or absent in intra-articular osteoid osteoma. A high level of suspicion is often required, especially when the nidus is small and associated reactive bone changes are minimal.
37,
38Occurrence in an atypical anatomic location may complicate the diagnosis of osteoid osteomas. Spinal osteoid osteoma, most commonly occurring in the neural arch of the lumbar spine, presents with scoliosis and radicular pain. CT or MRI is required in most cases to detect the nidus in the pedicle and lamina. Imaging and clinical findings of osteoid osteoma in the carpal and tarsal bones may mimic infection or inflammatory arthritis.
The differential diagnosis of an osteoid osteoma includes stress fracture, intracortical abscess, and other tumors such as intracortical hemangioma, osteoblastoma, and compensatory hypertrophy of the pedicle. Osteoid osteoma has a round nidus, as opposed to a stress fracture, which has a linear cortical break in the center of the cortical thickening. On bone scintigraphy, an osteoid osteoma demonstrates the “double density” sign with intense uptake in the nidus and surrounding moderate uptake. In contrast, a stress fracture typically shows linear intense uptake of the tracer.
39 An intracortical osteoid osteoma has a smooth margin with strong enhancement of the nidus; this contrasts with an intracortical abscess or sequestrum, which usually has an irregular shape and margin with peripheral rim enhancement at contrast-enhanced CT.
40 Differentiation between osteoid osteoma and intracortical hemangioma is difficult based on imaging features, although intracortical hemangioma is extremely rare. Compensatory hypertrophy of the vertebral pedicle is seen in unilateral spondylolysis and mimics spinal osteoid osteoma. Lack of a nidus and presence of contralateral spondylolysis in compensatory hypertrophy of the pedicle can differentiate it from spinal osteoid osteoma. The differences between osteoid osteoma and osteoblastoma are discussed in the following section.
Grossly and microscopically, osteoid osteoma shows a nidus rich in osteoblasts and poorly mineralized woven bone (
Fig. 23.10). A variably sclerotic bony rim surrounds the nidus. The current treatment of choice for osteoid osteoma is complete excision, which is curative with en bloc resection. Recently, percutaneous radiofrequency ablation of the nidus has become a treatment of choice almost replacing surgical excision. Radiofrequency ablation effectively relieves pain in most cases with few complications and a low recurrence rate.
41
Osteoblastoma
Osteoblastoma is a rare benign bone-forming neoplasm. It is characterized by loose woven bone bordered by prominent osteoblasts with reactive bone formation. Osteoblastoma is relatively rare, accounting for 3% of all benign bone tumors.
4 It affects patients in the second to fourth decades of life with male predilection (2.5:1).
42Histologically, osteoblastomas are identical to osteoid osteomas, but they are quite different regarding imaging findings and clinical manifestations. Compared to osteoid osteoma, osteoblastoma is less painful and less responsive to medication. Osteoblastoma is more expansile, larger (often arbitrarily distinguished from an osteoid osteoma by a measurement of more than 2 cm; mostly in the 3 to 10 cm range), and has less reactive adjacent bone change.
43 Unlike osteoid osteomas that more commonly involve the long tubular bones, osteoblastomas are most frequently found in the spine or flat bones
43; long tubular bone involvement is seen in 35% of cases, mostly in the diaphysis.
Radiographic features of osteoblastomas are nondiagnostic in most cases; the lesions may appear purely osteolytic or purely osteosclerotic or may demonstrate a combination of the two. In the long tubular bones, osteoblastomas may have an intramedullary or cortical origin. The appearance is usually that of an expansile, predominantly osteolytic lesion with an ossified matrix and reactive sclerosis. While CT is best for showing the calcified matrix and outer bony cortex (
Fig. 23.11), MRI is excellent at discriminating the tumor from associated marrow edema and showing soft tissue changes. Osteoblastomas are highly vascularized and demonstrate a dense capillary blush on angiogram. Bone scintigraphy can help pinpoint the tumor site, especially in those occurring in the posterior vertebral column, and shows increased uptake of the radionuclide in the lesion.
44Some osteoblastomas, in particular those arising from the posterior elements of the spine, have an ABC-like appearance. In other locations, the appearance of osteoblastoma is variable, and the differential diagnosis includes osteoid osteoma, SBC, ABC, eosinophilic granuloma, enchondroma, chondromyxoid fibroma, and fibrous dysplasia.
Curettage or en bloc surgical resection is the current management of choice for osteoblastomas.
Fibrous Cortical Defect/Nonossifying Fibroma
Fibrous cortical defects (FCDs) and nonossifying fibromas (NOFs) are benign fibrous lesions of bone and are histologically identical to each other. FCD is confined to the cortex of the bone and is smaller than 2 cm in size. NOF, also known as fibroxanthoma, has an eccentric location with medullary extension and is equal or larger than 2 cm in size. FCD is essentially a normal variant and is seen in children during skeletal maturation at age 4 to 8 years. Most (>70%) NOFs occur in teenagers.
4 FCD and NOF are the most common benign bone lesions, seen in up to 40% of children.
4FCDs are typically asymptomatic and detected incidentally; they spontaneously regress in most cases. NOFs are asymptomatic in most cases, but can present with pain associated with pathologic fractures. Although most NOFs present as a solitary lesion, there have been reported cases of multiple NOFs. Multiple NOFs along with café-au-lait skin lesions and other extraskeletal congenital malformations (mental retardation, ocular anomalies, cardiovascular malformations, hypogonadism) are known as Jaffe-Campanacci syndrome.
45 Some authors have proposed that it could be a different manifestation of neurofibromatosis type 1.
46,
47Radiography is usually sufficient for the diagnosis of FCDs and NOFs. Both FCDs and NOFs most commonly develop in the metaphyses of the long bones of the lower extremities with a predilection for the posterior cortex. FCD and NOF demonstrate a well-defined and cortex-based (FCD) or eccentrically located (NOF) osteolytic lesion with an adjacent sclerotic rim. In general, the lesions are not accompanied by substantial periosteal reaction. The larger lesions tend to have a more elongated and multiloculated appearance with slight expansion and cortical thinning (
Fig. 23.12). Main differential diagnoses include chondromyxoid fibroma, fibrous dysplasia, osteoid osteoma, bone abscess, and periosteal chondroma. FCDs and NOFs located in the metaphyses appear to migrate into the diaphysis with skeletal growth.
CT and MRI are not usually indicated, although CT may be useful in the cases of associated fracture. On MRI, the lesions are well-demarcated, typically in eccentric cortical locations, with low signal intensity on both T1- and T2-weighted MR images with enhancement depending on the evolving stage. As expected, an early active lesion appears hyperintense on T2-weighted MR images and enhances, while a regressing lesion shows a low signal on T2-weighted MR images with a lack of enhancement.
48 Whereas most NOFs can heal spontaneously with reactive sclerosis, some may persist into adulthood with a potential risk of developing a pathologic fracture.
Microscopically, FCD and NOF lesions are characterized by fibroblasts that are arranged in a storiform pattern, along with interspersed multinucleate giant cells and histiocytes (
Fig. 23.13). Hemosiderin and lipid may accumulate within the histiocytes. No treatment is required for noncomplicated NOF or FCD.
Fibrous Dysplasia
Fibrous dysplasia is a benign fibroosseous lesion involving the medullary cavity of the bone. It affects children and young adults with an equal gender distribution. Fibrous dysplasia has monostotic and polyostotic forms; the monostotic type accounts for 70% to 80% of cases.
49 The facial bones, in particular the jaw bone, are most commonly affected; other affected sites include the skull, ribs, and long bones including the femur and tibia. The polyostotic form, also known as fibrocartilaginous dysplasia or generalized fibrocystic disease of bone, can be associated with syndromes, such as McCune-Albright syndrome (precocious puberty, cutaneous café-au-lait lesions, unilateral polyostotic fibrous dysplasia) (
Fig. 23.14)
50,
51 and Mazabraud syndrome (intramuscular myxomas, fibrous dysplasia).
52On radiographs and CT, fibrous dysplasia demonstrates characteristic findings of a well-demarcated intramedullary lesion in a long bone with a ground-glass or hazy matrix. There is also endosteal scalloping with or without bone expansion (
Fig. 23.14). “Shepherd crook deformity” is a result of repeated fractures and varus deformity of the proximal femur. There is no periosteal reaction unless it is fractured. Bone scintigraphy may help identify multiple lesions. On MRI, fibrous dysplasia has similar signal intensity to that of muscle on T1-weighted MR images and variable signal on T2-weighted MR images depending on the component (although showing T2 hyperintensity in most cases). After administration of gadolinium-based contrast material, the lesions usually enhance heterogeneously (
Fig. 23.15).
53Microscopically, fibrous dysplasia shows delicate spindled to stellate fibroblasts with variable amounts of admixed thin bony trabeculae without substantial osteoblastic rimming (
Fig. 23.16). The prognosis for this benign disease is excellent.
However, affected pediatric patients can occasionally be left with deformities such as leg length discrepancy or bowing. Malignant transformation is exceptionally rare.
Osteofibrous Dysplasia
Osteofibrous dysplasia is a benign fibroosseous lesion of bone. It is confined almost exclusively to the tibia. It occurs mostly during infancy and childhood and is rare after the age of 15 years. Osteofibrous dysplasia shares imaging features with adamantinoma, which has a progressive and malignant nature.
54,
55 It has been suggested that osteofibrous dysplasia may represent a precursor to, an incomplete sampling of, or a regression of adamantinoma.
56On radiographs, osteofibrous dysplasia is typically seen as a relatively well-circumscribed complex radiolucent lesion with marginal sclerosis involving the anterior cortex of the tibia. There is typically associated cortical thickening and anterior bowing (
Fig. 23.17). The eccentric cortical location differentiates it from fibrous dysplasia, which has a medullary location.
In contrast to adamantinoma, which often has a soft tissue component, osteofibrous dysplasia typically is not accompanied by a soft tissue mass. Pathologic fractures and congenital pseudoarthrosis can be associated with osteofibrous dysplasia. CT and MRI show the extent of the lesions and the degree of narrowing of the medullary cavity as well as the thinned outer cortex. MRI signal intensity of osteofibrous dysplasia is not specific; it is of low to intermediate signal on T1-weighted MR images and high signal on T2-weighted MR images with a varying degree of sclerosis seen as a low signal rim. There is usually strong intralesional enhancement after contrast administration (
Fig. 23.17).
Microscopically, osteofibrous dysplasia contains delicate fibroblasts that produce wispy collagen (
Fig. 23.18). Bony trabeculae are embedded within the fibrous proliferation. Occasional keratin-positive cells, occurring singly and not well visualized on routine stains, may be highlighted by immunohistochemical stains. Most osteofibrous dysplasias undergo spontaneous healing or regression. In rare cases, they may progress to adamantinoma.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH), previously known as eosinophilic granuloma or histiocytosis X, is a neoplastic proliferation of Langerhans cells. LCH is a spectrum of histiocytic disorders in which immature Langerhans cells proliferate in certain parts of the body. The skeletal system is affected in up to 80% of cases.
4 LCH may occur at any age but is mostly seen under the age of 5 years.
57 Systemic involvement is seen in infants and younger pediatric patients. The prognosis is poor in children younger than 2 years.
Traditionally, LCH was divided into three categories according to clinical manifestations: eosinophilic granuloma (skeleton solely involved), Hand-Schuller-Christian disease (cranial lesions, diabetes insipidus, and exophthalmos), and Letterer-Siwe disease (disseminated form that occurs in infants and very young children). More practically, it can be categorized into a restricted (monostotic or polyostotic) or an extensive (visceral organ involvement) form. Clinical symptoms vary with involved organs, and affected pediatric patients often present with low-grade fever and raised inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein.
The skull is most frequently involved, although any of the axial or appendicular bones can be affected by LCH. A punched-out lytic lesion with a beveled edge is characteristic of the skull lesions. Beveled edges are explained by differential destruction of the inner and outer table of the skull (
Fig. 23.19). When LCH involves the maxilla or mandible, it can result in “floating teeth.” This is the description given to the appearance on imaging of teeth “hanging in the wind” as a result of underlying alveolar bone destruction around the root of the teeth in patients with
LCH. LCH lesions of the mastoid bone cause ear symptoms. Therefore, the affected children are prone to treatment for otomastoiditis before being diagnosed with LCH. LCH is the most common cause of “vertebra plana” in children (
Fig. 23.19). For spinal LCH lesions, MRI is needed to assess the presence of an epidural mass and cord compression. In long tubular bones, the lesions typically develop in the metaphysis or diaphysis. LCH involving uncommon locations such as the clavicle and small tubular bones has also been reported.
58A skeletal survey is the first step to be used to investigate bone involvement of LCH despite some controversy regarding its sensitivity in comparison with bone scintigraphy, which is a radiation bearing.
59,
60 In children with multiple bone lesions, LCH should be included in the diagnostic considerations. Although radiological findings vary with the stage and disease activity, most LCH bone lesions are well-defined (“punched out”) and purely osteolytic with endosteal scalloping (
Fig. 23.19). Sometimes, a permeative and aggressive pattern of bone destruction is present in association with either a single or multilayered periosteal reaction (
Fig. 23.20), which may mimic findings of malignant bone tumors or osteomyelitis. Spontaneous regression is not
unusual in LCH, and involuting lesions appear more sclerotic and remodeled.
Cortical breakdown and soft tissue extension of LCH can be well demonstrated on CT; the margins are irregular and tend to be more sharply delineated as compared to those in Ewing sarcoma. On MRI, the bone lesions and soft tissue masses of LCH appear with low signal intensity on T1-weighted MR images, high signal intensity on T2-weighted MR images, and enhance with gadolinium. A soft tissue mass is present in about one-third of lesions. The associated inflammatory reaction causes bone and soft tissue edema, which enhance and may mimic osteomyelitis (
Fig. 23.20). Involuting lesions can show low signal intensity on both T1- and T2-weighted MR images. WB MRI using the STIR sequence can provide valuable information in the initial workup as well as for patient monitoring and has many advantages over radiography and bone scintigraphy.
61Pathological evaluation of LCH shows sheets of Langerhans-type “histiocytes” as well as variable numbers of admixed eosinophils. LCH localized to the skeletal system carries a favorable prognosis. “Vertebra plana” tends to restore its height, and the involved bones remodel themselves after treatment. Reactivations are relatively common, occurring in about a quarter of the patients with systemic disease. LCH is treated by low-dose chemotherapy in extensive cases.
Chondroblastoma
Chondroblastoma is a benign cartilaginous neoplasm that affects exclusively the epiphyses or apophysis of long bones. It is most common in the second and third decades of life, and almost half of the lesions occur prior to physeal closure.
62 Chondroblasts, chondroid matrix, giant cells, and foci of calcifications are seen on microscopic examination. Chondroblastomas can have a component of ABC. There are two main benign bone lesions that affect the epiphyses: chondroblastoma prior to skeletal maturation and GCT after physeal closure
(Schematic B). The differential diagnosis for an epiphyseal lesion in a child can also include infectious osteomyelitis (Brodie abscess), which was discussed in
Chapter 22.
Chondroblastoma typically presents as an eccentric well-defined osteolytic lesion with a sclerotic rim in the epiphysis or apophysis (epiphysis equivalent) on radiographs (
Fig. 23.21). Approximately one-third of cases show calcified chondroid matrix, which is best shown by CT
63,
64. Surrounding inflammatory changes are usually pronounced, resulting in adjacent bone marrow and soft tissue edema. Periosteal reaction is common due to an associated inflammatory response.
63 In contrast to most cartilaginous tumors, which are usually hyperintense on T2-weighted MR imaging, chondroblastoma can present with low- to intermediate-signal intensity on T2-weighted MR images. An ABC component can show cystic areas with or without fluid-fluid levels (
Fig. 23.21). Chondroblastoma shows a low signal intensity rim on MRI. The bone marrow edema, soft tissue changes, and/or joint effusions can be pronounced. These findings can be important for differentiating chondroblastoma from other benign tumors. Chondroblastomas variably enhance, while Brodie abscesses typically show enhancement of the granulation tissue around the nonenhancing central abscess. The various signal intensities on T2-weighted MR imaging and enhancement patterns of chondroblastomas correlate well with their underlying histopathologic features, depending on the various amounts of chondroid matrix, calcifications, ABC components, and others.
64Microscopically, chondroblastomas show sheets variably mature chondrocytes, often individually outlined by a rim of calcium (so-called “chicken-wire calcification”). Admixed osteoclast-like giant cells are frequently seen. Simple curettage
is curative in the majority of patients, although recurrence may be observed in 14% to 18%.
65
Chondromyxoid Fibroma
Chondromyxoid fibroma is a relatively rare benign cartilaginous tumor that usually involves the metaphyses of the extremity bones.
66 The proximal tibia is the most common location, followed by the fibula and the calcaneus.
67 Lesions involving the upper extremity bones, pelvis, and spine have also been described. It is more common in males in the second or third decades of life.
68On radiographs, chondromyxoid fibroma shows an eccentric osteolytic lesion with a sclerotic margin in the metaphysis; its elongated shape parallels the axis of the long tubular bone. In small tubular bones, it can occupy the entire width of the medullary cavity with bony expansion and cortical thinning (
Fig. 23.22). Calcifications are not usually present. MR signal characteristics are nonspecific and vary depending on the internal composition, but usually show low signal on T1- and high signal on T2-weighted MR images (
Fig. 23.22).
The current management of choice for chondromyxoid fibroma is surgical resection. Chondromyxoid fibromas can recur after surgical excision, but there is no known risk of malignant transformation.
Enchondroma
Enchondroma is a type of chondroma that involves the medullary cavity. It is composed of benign hyaline cartilage.
Chondromas may also occur in a juxtacortical or periosteal location. The peak age of juxtacortical chondromas is slightly younger than that of enchondromas.
69 Enchondromas commonly occur in the metaphyses or metadiaphyses of the small tubular bones of the hands and feet of children and adults. Enchondroma protuberans has been described as a variant of enchondroma that protrudes outward from one side of the affected bone mimicking an osteochondroma or juxtacortical chondroma.
64Ollier disease and Maffucci syndrome are the most common subtypes of endochondromatosis, in which enchondromas can affect both sides of the body, but are random and asymmetric; generally, one side of the body is almost exclusively or more predominantly affected.
70 Enchondromas that arise in the metaphysis near the growth plates may impair physeal bone growth resulting in deformity and limb shortening. The risk of malignant transformation to chondrosarcoma is also a potential complication. Maffucci syndrome refers to multiple enchondromas in association with spindle cell hemangioma (
Fig. 23.24). Both Ollier disease and Maffucci syndrome are known to be nonhereditary, usually sporadic disorders. The enchondromas and spindle cell hemangiomas usually harbor somatic mutations in the IDH1 or IDH2 gene.
71Radiographic evaluation is often sufficient to make a diagnosis of enchondroma. Typical radiographic findings include multiple expansile, radiolucent lesions with well-defined bony margins in the metaphyses of long bones and small tubular bones. Enchondromas develop in close proximity to the growth plate and then migrate toward the diaphysis (
Figs. 23.23 and
23.24). In the long bones, they commonly show longitudinal streaks in the metaphysis that parallel the long bone axis. Metaphyseal widening by clustered enchondromas and variable shortening of metacarpal or phalangeal bones are seen in the hands (
Figs. 23.23 and
23.24). On both radiography and CT, punctuate or chondroid calcifications can be seen. On MRI, signal intensity follows that of the cartilage at all sequences, revealing predominantly high signal intensity on T2-weighted MR images. Enhancement after gadolinium administration varies, and some lesions show enhancement in a peripheral, ring-and-arc pattern.
Giant Cell Tumor of Bone
GCT is a benign but locally aggressive bone tumor. It accounts for 4% to 5% of all primary bone tumors and usually affects patients from 20 to 45 years of age with a female predilection.
73,
74 Uncommonly, younger pediatric patients are affected. The tumor typically involves the long tubular bones, specifically the distal femur and proximal tibia. Spinal involvement is described in a mainly adult population.
75 Although GCT is rare in skeletally immature patients, GCT can develop in the distal metaphysis and involve the epiphysis before the physis is closed. The tumor is located at the epimetaphysis in most cases, and tumor extension through growth plates may reflect its aggressiveness. Purely metaphyseal GCT has been observed only in the distal radius.
76 Pulmonary “ metastasis” occurs rarely; it is indolent, thought to represent tumor emboli rather than true metastasis.
Radiographically, GCT is typically a geographic, expansile, osteolytic lesion with ill-defined or sclerotic margins seen in the epimetaphysis of the tubular bones (
Fig. 23.26). Matrix calcification is not a finding in GCT. In cases of expansile bone lesions, there can be cortical breakdown with soft tissue extension. No periosteal reaction is identified unless complicated by a pathologic fracture. MRI shows diverse findings depending on whether the tumor is mainly solid or cystic. A solid tumor shows intermediate signal intensity on both T1- and T2-weighted MR images with diffuse enhancement. There can be associated intratumoral hemorrhage and intratumoral ABC components. Fluid-fluid levels can be demonstrated on both CT and MRI.
Microscopically, GCT of bone shows numerous large multinucleate giant cells embedded in a background of mononuclear fibrohistiocytic cells (
Fig. 23.27). En bloc resection is preferred over curettage due to the high recurrence rate of the latter procedure. Local recurrent is not common.








































 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access