In this article, the genotype-MR phenotype correlation of the most common or clinically important inherited metabolic diseases (IMD) in the pediatric population is reviewed. A nonsystematic search of the PubMed/Medline database of relevant studies about “genotype-phenotype correlation” in IMD was performed. Some MR phenotypes related to specific gene mutations were found, such as bilateral hypertrophy of inferior olives in patients harboring POLG and SURF1 mutations, and central lesions in the cervical spinal cord in patients with nonketotic hyperglycinemia harboring GLRX5 gene mutation.
Key points
- •
Inherited metabolic diseases (IMD) form a group of disorders, mostly genetically inherited, caused by abnormal protein formations.
- •
One strategy to increase the accuracy of neuroimaging in IMD is to compare the genetic information (genotype) with the imaging phenotype detected by MRI (MR phenotype).
- •
Some MR phenotypes are related to specific gene mutations, such as bilateral hypertrophy of inferior olives in patients harboring POLG and SURF1 mutations, and central lesions in the cervical spinal cord in nonketotic hyperglycinemia patients harboring GLRX5 gene mutation.
- •
From a neuroimaging point of view, a desirable scenario of the future could be using brain imaging, such as advanced MRI, as a biomarker (MR phenotype) to predict the most probable genetic abnormality of a specific neurologic disease (genotype).
Introduction
Significant advances in imaging of the structure and function of the brain, brainstem, and cerebellum have been made in the last decades. High-resolution MRI and diffusion tensor imaging have been extensively used in both clinical and neurosciences settings, expanding and redefining the applications of modern neuroimaging techniques. An example of this issue can be clearly found in the malformation of the cerebral cortex, in which development of MRI technology associated with a better understanding of embryology, neurodevelopment, and neurogenetics has dramatically changed the way researchers classified these diseases.
Equal achievements occurred in our understanding of the human genome and its role in normal and abnormal development of the central nervous system (CNS). From a biological perspective, the identification of a genetic abnormality can improve our understanding of the mechanism of specific diseases. From a clinical perspective, a genetic diagnosis could optimize diagnosis, prognosis, and treatment of neurologic disorders.
In this context, imaging and genetic studies have been predominant in the investigation of many pediatric neurologic disorders, particularly congenital malformations of the CNS (CMCNS) and inherited metabolic disorders (IMD). Linking genetic data and neuroimaging phenotype is an emerging approach in neuroscience to better understand the complex imaging appearance of these disorders. From a neuroimaging point of view, a desirable scenario of the future could be using brain imaging, such as advanced MRI, as a biomarker (MR phenotype) to predict the most probable genetic abnormality of a specific neurologic disease (genotype). This approach could be useful to both neuroscientists (better genetic-neuroimaging integration) and clinical physicians (better approach to neurologic diseases), making imaging a better diagnostic tool for more effective treatment and prognosis definition.
In this article, the genotype-MR phenotype correlation of the most common or clinical relevant IMD and CMCNS in the pediatric population are reviewed. Although many disorders could be included in this review, the data focus on the most commonly diagnosed diseases and those in which the neuroimaging abnormalities are better defined. The PubMed/Medline database was searched with a combination of key words such as “genotype-phenotype correlation,” “genotype-imaging correlation”, and “neuroimaging-genetic correlation” in both IMD and CMCNS. The searches were limited to articles in English and those with abstracts. After the searches were conducted, the abstracts of the returned articles were examined, to determine their applicability for review. Relevant studies were defined liberally to be those that included any discussion about correlation of neuroimaging and a genetic abnormality in both IMD (part 1) and CMCNS (part 2 elsewhere in this issue). A review of the general rules of genotype-phenotype correlation is included in this review (part 1), because we believe that knowledge of the principles of medical genetics is essential for every neuroscientist (including radiologists).
Introduction
Significant advances in imaging of the structure and function of the brain, brainstem, and cerebellum have been made in the last decades. High-resolution MRI and diffusion tensor imaging have been extensively used in both clinical and neurosciences settings, expanding and redefining the applications of modern neuroimaging techniques. An example of this issue can be clearly found in the malformation of the cerebral cortex, in which development of MRI technology associated with a better understanding of embryology, neurodevelopment, and neurogenetics has dramatically changed the way researchers classified these diseases.
Equal achievements occurred in our understanding of the human genome and its role in normal and abnormal development of the central nervous system (CNS). From a biological perspective, the identification of a genetic abnormality can improve our understanding of the mechanism of specific diseases. From a clinical perspective, a genetic diagnosis could optimize diagnosis, prognosis, and treatment of neurologic disorders.
In this context, imaging and genetic studies have been predominant in the investigation of many pediatric neurologic disorders, particularly congenital malformations of the CNS (CMCNS) and inherited metabolic disorders (IMD). Linking genetic data and neuroimaging phenotype is an emerging approach in neuroscience to better understand the complex imaging appearance of these disorders. From a neuroimaging point of view, a desirable scenario of the future could be using brain imaging, such as advanced MRI, as a biomarker (MR phenotype) to predict the most probable genetic abnormality of a specific neurologic disease (genotype). This approach could be useful to both neuroscientists (better genetic-neuroimaging integration) and clinical physicians (better approach to neurologic diseases), making imaging a better diagnostic tool for more effective treatment and prognosis definition.
In this article, the genotype-MR phenotype correlation of the most common or clinical relevant IMD and CMCNS in the pediatric population are reviewed. Although many disorders could be included in this review, the data focus on the most commonly diagnosed diseases and those in which the neuroimaging abnormalities are better defined. The PubMed/Medline database was searched with a combination of key words such as “genotype-phenotype correlation,” “genotype-imaging correlation”, and “neuroimaging-genetic correlation” in both IMD and CMCNS. The searches were limited to articles in English and those with abstracts. After the searches were conducted, the abstracts of the returned articles were examined, to determine their applicability for review. Relevant studies were defined liberally to be those that included any discussion about correlation of neuroimaging and a genetic abnormality in both IMD (part 1) and CMCNS (part 2 elsewhere in this issue). A review of the general rules of genotype-phenotype correlation is included in this review (part 1), because we believe that knowledge of the principles of medical genetics is essential for every neuroscientist (including radiologists).
Genotype-phenotype correlation
In neurosciences, few topics have advanced as fast as neurogenetics. Data from the last 20 years in recombinant DNA technology and polymerase chain reaction (PCR) linkage studies significantly increase the number of neurologic disease genes identification, but identifying genes for both autosomal dominant and recessive diseases has been challenging. However, the advent of next-generation sequencing (NGS) with the whole genome sequencing (WGS), target sequencing, and whole exome sequencing (WES) has dramatically changed this scenario. These new techniques are emerging as a tool to facilitate cost-effective molecular diagnoses in routine clinical care of these disorders.
As a rule, most genes have been identified by defining a candidate gene by both its chromosomal location and its proprieties. Gene identification was based on methods in which the chromosomal location of the disease locus was not required (positional-independent strategies) and those that depended on this knowledge (positional cloning). In the former, gene identification was achieved based on thorough knowledge of the protein product, DNA sequence, or its normal function. In the latter, disease genes were identified using only knowledge of their approximate chromosomal location. Although both strategies are separate in principle, most commonly, geneticists and neuroscientists use a combination of both sets of information to identify a potential genetic variation. The most often used tool is based on NGS (as WGS and WES) and genome-wide association studies. There are some bioinformatics tools that help researchers to predict if a genomic variation could be damaged, but all variations should be tested with functional studies to ensure that they are related to a good candidate gene consistent with the disease phenotype.
In this context, the association between the presence of a certain mutation or mutations (genotype) and the resulting physical trait, abnormality, or pattern of abnormalities (phenotype) has been studied as a promising investigational tool. Several general principles have emerged as a result of the intensive study of causative variations in genetic disorders.
According to the Mendelian paradigm, a specific disease is caused by mutations in a single gene. Because there are 2 alternative forms of any given gene (alleles), a genetic disease can occur if a mutated allele was inherited from the father or from the mother (autosomal dominant transmission) or if both mutated alleles were inherited from the parents (autosomal recessive transmission). However, even single gene (monogenic) diseases can manifest with a wide range of symptoms, severity, and prognostic issues, which is called variable expressivity.
However, most neurologic disorders are complex (multifactorial) and not monogenic disorders. There are variations in the genome that are more frequent in the population (polymorphisms) and that can play a role in several common diseases, whenever they occur in a particular combination with an overall effect that causes a biochemical/biological pathologic condition. Environmental effects can also contribute to the pathophysiology of some neurologic diseases.
There are several limitations for the genotype-phenotype correlation. For example, mutations in the same gene may be responsible for more than 1 disease. In the same context, mutations in a single gene (allelic heterogeneity) can cause different and distinct clinical phenotypes and, on the other hand, there are many similar clinical phenotypes caused by mutations in different genes (nonallelic heterogeneity or genetic heterogeneity). Also, mutations in more than 1 gene may be required to express a given clinical phenotype (digenic inheritance or triallelic inheritance), and different mutations in the same gene may give rise to distinct dominant and recessive forms of the same disease. The definition of the phenotype is mostly qualitative, being either present or absent, which could be a confounder in establishing a genotype-phenotype correlation of a specific disease.
Neuroimaging in inherited metabolic disorders
IMD are a group of diseases caused by abnormal protein formations as a result of a gene mutation. These anomalous proteins disrupt the function of 1 or more metabolic pathways. The diagnosis of these disorders is a challenge, and usually, the clinical presentation is variable. As a result, most patients do not receive a diagnosis, despite the investigation. Although most IMD cannot be treated, a specific diagnosis is critical for some treatable metabolic diseases and also for proper genetic counseling.
Because of its high sensitivity in detecting changes in myelination, structural damage, and specific patterns of brain involvement, MRI techniques (structural imaging, diffusion imaging, and proton spectroscopy) are the modalities of choice to investigate patients with inborn errors of metabolism (IEM). However, imaging interpretation is complex. As a rule, it demands the knowledge of many radiologic signs, age of onset of the disease, genetic pattern, and recognition of the most important clinical findings.
Strategies to increase the accuracy of neuroimaging in IEM, such as systematic analysis of the pattern of CNS involvement and the discriminating MRI pattern approach, have been described. With recent advance in genetic and neuroimaging technology, it is becoming possible to compare the genetic information (genotype) with the imaging phenotype detected by MRI (MR phenotype). The goal is to identify specific MRI phenotypic changes related to a particular genetic abnormality. Although difficult to achieve, there is a potential benefit of this strategy to reduce the differential diagnosis, guide clinical decisions, and reduce cost of additional tests, including imaging and genetic tools.
MR phenotype-genotype correlation in disorders involving energy metabolism
This group includes disorders with clinical findings related to a deficiency in energy production or utilization, such as mitochondrial defects, disorders of glycolysis, glycogen metabolism, gluconeogenesis, and creatine metabolism. Most of the genotype-MR phenotype of this group is related to mitochondrial respiratory chain defect disorders.
The mitochondrion is a highly specialized organelle occurring in almost all eukaryotic cells producing cellular energy through oxidative phosphorylation (OXPHOS). Its role depends on a complex relationship between the mitochondrial genome (mtDNA) and the nuclear genome (nDNA), and the nDNA has a fundamental role for most of the OXPHOS system, for maintaining and replicating mtDNA and also for the organelle network proliferation and destruction. Almost 100 structural OXPHOS subunit genes have been identified, and approximately 80% are encoded by the nuclear genome, including the complex I-V subunits.
Defects in either the mtDNA or nDNA are responsible for mitochondrial diseases (a group of genetically and phenotypically diverse disorders). With a prevalence of 1 in 10,000 newborns, the diagnosis should be considered when a patient presents with a progressive course involving seemingly unrelated organs. A wide variety of neurologic manifestations such as optic atrophy, seizures, strokelike episodes, ataxia, and neuropathy can be present. A detailed family history is important; a clear maternal inheritance (without male transmission) indicates a primary mtDNA defect, whereas an autosomal inheritance pattern indicates nDNA interaction.
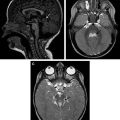
Neuroimaging of patients with mitochondrial diseases usually shows focal or diffuse lesions in the cerebral cortex, white matter, basal ganglia, and brainstem. Restricted diffusion and lactate peak on MR spectroscopy are also common ( Figs. 1 and 2 ). Cerebral and cerebellar atrophy appears in patients with longer disease duration. In many cases, blood or cerebrospinal fluid (CSF) lactate concentration, cardiac evaluation, and muscle biopsy for histologic or histochemical evidence can indicate mitochondrial disease. However, establishing a molecular genetic diagnosis is preferred.
Specific genes are known to be responsible for some mitochondrial respiratory chain defect disorders, although different mutations in the same gene can cause a variety of diseases ( Table 1 ). A genetic review of all genes related to mitochondrial disorders, including mutations associated with complex I-V deficiencies, is beyond the scope of this article, but a review of 1 gene (POLG) is useful to understand the complexity of this issue.
| Mutation Site | Disease or Syndrome |
|---|---|
| POLG (15q25) | KSS PEO Alpers disease Ataxia-neuropathy syndrome MNGIE (without CNS commitment) |
| SURF1 (3q34) | Leigh syndrome |
| TYMP (22q13.32) | MNGIE (with CNS commitment) |
| Multiple deletion of mtDNA | MELAS |
POLG (polymerase DNA directed γ) located on 15q23 region is the gene that codes for the catalytic subunit of the mtDNA polymerase. Mutations in the POLG gene are an important cause of pediatric mitochondrial disease, such as Kearns-Sayre syndrome (KSS) and Alpers syndrome. Studying patients with A467T and W748S POLG mutations, Tzoulis and colleagues reported lesions in the occipital cortex, deep cerebellar structures, and the thalamus, which spare the basal ganglia. An additional potential neuroimaging discriminator feature is a pattern of involvement of the inferior olivary nuclei. Kinghorn and colleagues studied 3 patients, 2 of whom were heterozygous (p.Ala467Thr and p.Trp748Ser) for POLG gene mutations. Both of them had bilateral signal abnormality and hypertrophy of the inferior olives. However, these alterations are not pathognomonic for POLG mutations, because they were also described in patients with SURF1 gene mutations with Leigh syndrome (LS) phenotype. Also, at least 150 mutations (see http://www.hgmd.org ) in the POLG have been described and were associated with several phenotypes of neurodegenerative diseases.
Leber Hereditary Optic Neuropathy
Leber hereditary optic neuropathy (LHON) is a common cause of inherited blindness, which typically presents with bilateral, painless, subacute visual failure in young adult men. Clinical diagnosis is usually confirmed by molecular genetic analysis showing a mtDNA mutation affecting genes coding for complex I subunits of the respiratory chain, such as m.3460G>A, m.11778G>A, and m14484T>C. MRI is usually normal or shows increased T2 signal changes in the optic tract and chasm. Swelling and enhancement of the optic nerves/chiasm on gadolinium-enhanced imaging with signal changes in the optic tracts have been described. LHON associated with cerebellum and inferior olivary nucleus atrophy can suggest a specific mtDNA pattern related to G11778A and T3394C mutations.
Leigh Syndrome
LS, also known as subacute necrotizing encephalomyelopathy, refers to a symptom complex, characterized by progressive neurodegeneration, with variable clinical and pathologic manifestations. The classic pathologic abnormalities are microcystic cavitation, vascular proliferation, neuronal loss, and demyelination in the midbrain, basal ganglia, cerebellar dentate nuclei, and cerebral white matter.
Several mutations of both mtDNA and nDNA are responsible for the LS, involving genes coding for proteins in respiratory chain complexes I, II, III, IV, and V, mitochondrial transfer RNA (tRNA), pyruvate dehydrogenase complex, and coenzyme Q10. All of them were divided into 4 groups: defects of the pyruvate dehydrogenase complex; cytochrome oxidase deficiency; patients with apparently mutation on the mitochondrial adenosine triphosphatase 6 gene; and complex I deficiency. The group of cytochrome C oxidase complex (respiratory chain complex IV) deficiency is believed to be the most common cause of LS.
Studies have tried to identify a homogeneous neuroimaging pattern associated with SURF1 mutations. The characteristic lesion distribution of patients harboring any mutation was along the brainstem, thalamus, and basal ganglia. However, it seems that LS caused by SURF1 mutations presents a peculiar MRI pattern of bilateral involvement of the subthalamic nuclei. Farina and colleagues performed a study with 22 patients with LS, 8 with SURF1 mutations and 14 with mutations in other genes. All of the patients with SURF1 mutations presented subthalamic nuclei abnormality, compared with 2 patients in the other group. These results were confirmed in other studies shown in Table 2 . The involvement of the basal ganglia, which used to be considered the hallmark of LS, is not essential for the diagnosis. The basal ganglia spared seem to be more common in patients harboring SURF1 mutations.
| Studies | Total Number of Patients | MRI Findings (Number of Patients with Affected Brain Region) | ||||
|---|---|---|---|---|---|---|
| Brainstem | Cerebellar | Subthalamic Nuclei | Thalami | Basal Ganglia | ||
| Farina et al, 2002 | 8 | 8 | 6 | 8 | 2 | 2 |
| Xie et al, 2009 | 8 | 3 | 3 | 3 | 1 | 2 |
| Rossi et al, 2003 | 3 | 3 | 3 | 3 | 0 | 1 |
| Sonam et al, 2014 | 4 | 4 | 4 | 0 | 0 | 2 |
| Bruno et al, 2002 | 1 | 1 | 0 | 1 | 0 | 0 |
| Salviati et al, 2004 | 1 | 1 | 1 | 0 | 0 | 0 |
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes
Mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) refers to a group of disorders with recurrent strokelike events, with either permanent or reversible neurologic deficits. The classic presentation is strokelike cortical lesions (hyperintense on T2/fluid-attenuated inversion recovery [FLAIR] images with restricted diffusion), crossing vascular territories with shifting spread (appearance, disappearance, reappearance elsewhere). The involvement of the parieto-occipital lobe is usually more extensive, and intracranial calcification (usually in the basal ganglia) is also common, as shown in Table 3 .
| Studies | CT and MRI Findings (Affected Brain Region) | ||||
|---|---|---|---|---|---|
| BG Calcification | Hyperintensity Lesion in T2 | ||||
| Occipital | Temporal | Parietal | Frontal | ||
| Bi et al, 2006 | + | + | + | + | − |
| Karkare et al, 2009 | − | + | + | + | − |
| Chung et al, 2005 | + | + | + | + | − |
| Liu et al, 2013 | + | + | + | − | − |
| Casimiro et al, 2012 | 0 | − | + | + | − |
| Total | 3/5 | 4/5 | 5/5 | 4/5 | 0/5 |
MELAS is caused by mtDNA mutations, especially in the MTTL1 gene, which encodes tRNAleu in about 80% of the cases. The most common mutation is m.3243A>G. In 1998, Sue and colleagues studied 26 patients with MELAS, 22 of whom harbored the m.3243A>G mutation. The most common neuroimaging finding was basal ganglia calcification (seen in both young and adult patients), which was present in 14 patients (11 with the m.3243A>G mutation). Globus pallidus was seen in 12 patients.
Alpers Disease
Alpers disease is an autosomal recessive disorder characterized by rapid progressive cerebral and hepatic degeneration caused by mutations in the POLG gene. CNS compromise is characterized by progressive degeneration of the cerebral and cerebellar cortex, thalamus, and basal ganglia, usually in the first years of life. MRI of patients with Alpers can be normal in the early stages, but the classic imaging findings are T2/FLAIR hyperintensity in the cortex, subcortical white matter, and basal ganglia, with cortical thinning, especially in the frontal, posterior temporal, and occipital lobes. In a recent retrospective study of 66 children with mitochondrial disorders, 10 patients with Alpers disease were evaluated. All of them had POLG mutations (p.A467T/ R574W, p.A467T/G303R, and p.W748S–7p.Q497H–4p.E1143G/ R852C). The most prevalent finding was cerebral cortex atrophy, which presented in 70% of the patients. Basal ganglia involvement, considered a classic feature, was presented just in 30%.
Progressive External Ophthalmoplegia and Kearns-Sayre Syndrome
Progressive external ophthalmoplegia (PEO) can appear isolated or in association with KSS. In addition to PEO, KSS consists of retinal dystrophy and at least 1 of the following features appearing before 20 years old: heart block, cerebellar ataxia or increased protein in CSF. MRI of patients with PEO and KSS is similar, showing T2/FLAIR hyperintensities in the white matter, with early involvement of the subcortical U fibers and sparing of periventricular areas. Later in the course of the disease, deep cerebral white matter and the deep gray matter nuclei, particularly the dorsal midbrains, medial and posterior thalami, and the globus pallidus, show T2 hyperintesity ( Fig. 3 ). PEO and KSS can be caused by POLG mutations, but other genes, such as PEO1, OPA1, and RRM2B, are associated with this condition.