Bronchiectasis
Etiology
Diseases of the airways are surprisingly common in clinical practice, and imaging tests have a central role in the evaluation of the patient. Bronchiectasis is an important yet, in historic terms, a surprisingly neglected disorder characterized pathologically by abnormal permanent dilatation of the bronchi, leading to significant morbidity and mortality. The causes and associations of bronchiectasis are diverse and summarized in Box 57.1 . However, the relative impact of the various etiologic factors, such as infection, has changed over time, and there is considerable variation dependent on geographic and ethnic factors. The availability of effective antimicrobial therapy and the policy of widespread childhood immunization in many countries have had a significant impact, leading not only to a decrease in the prevalence of postinfectious bronchiectasis but also to a decline in overall mortality.
Idiopathic
Congenital
Cystic fibrosis
Mounier-Kuhn syndrome (tracheobronchomegaly)
α1-Antitrypsin deficiency
Williams-Campbell syndrome
Intralobar sequestration
Postinfectious or postinflammatory
Bacterial, mycobacterial, viral, protozoal
Swyer-James (MacLeod) syndrome
Aspiration of gastric contents
Inhalation of toxic fumes
Immunodeficiency
Primary: selective immunoglobulin deficiency, panhypogammaglobulinemia
Secondary: malignant neoplasia, chemotherapy
Mechanical airway obstruction
Inhaled foreign bodies
Neoplastic
Broncholithiasis
Bronchostenosis
Defective mucus transport
Primary ciliary dyskinesia
Secondary ciliary dyskinesia
Young syndrome
Immunologic
Allergic bronchopulmonary aspergillosis
After heart-lung, lung, or bone marrow transplantation
Miscellaneous associations
Middle lobe syndrome
Rheumatoid arthritis
Sjögren syndrome
Systemic lupus erythematosus
Ulcerative colitis
Yellow nail syndrome
Celiac disease
Human immunodeficiency virus infection
In the developed world, including the United States and many countries in Europe, postinfectious bronchiectasis is now less of a clinical problem; the more common causes include cystic fibrosis and immunologic deficiencies, which may be primary (as seen in patients with panhypogammaglobulinemia or more selective immunoglobulin subclass defects) or secondary to immunosuppression (as may be seen in the context of hematologic malignant disease or human immunodeficiency virus–related disease). In stark contrast, infections such as tuberculosis continue to account for the majority of patients with bronchiectasis in developing countries. Defects of mucociliary clearance (either congenital, as in primary ciliary dyskinesia, or acquired), mechanical obstruction (intrinsic or extrinsic), and congenital abnormalities (such as intralobar sequestration) are among the less common but recognized causes of bronchiectasis. However, a cause for bronchiectasis may not be identified in 30% to 70% of patients. Another complicating factor in the evaluation of patients with bronchiectasis is that in a small proportion of cases, more than one causative factor may be identified.
Prevalence and Epidemiology
The true prevalence of bronchiectasis is unknown. Because it has long been the paradigm of an “orphan” lung disease, reliable published data on the prevalence of bronchiectasis have been frustratingly difficult to come by. One of the fundamental problems has stemmed from the relatively widespread misconception that bronchiectasis is no longer a significant clinical entity. The corollary is that the index of clinical suspicion for a diagnosis of bronchiectasis has been low, and physicians often attribute the symptoms and signs of bronchiectasis to other respiratory diseases (e.g., smoking-related chronic bronchitis) that are perceived to be more common.
Nevertheless, the prevalence of bronchiectasis is known to be remarkably high in select populations; for instance, high rates of bronchiectasis are known to be a problem in indigenous Australians and also native Alaskans. Similarly, bronchiectasis unrelated to cystic fibrosis is strikingly prevalent in parts of New Zealand, where there is an estimated overall prevalence of 1 in 3000 in children younger than 15 years and a staggering prevalence of 1 in 625 for children of Pacific ancestry. These data in contrast to the comparatively low prevalence of bronchiectasis in Finnish children of a similar age. Finally, in most large series of patients with bronchiectasis, a consistent finding has been the preponderance of female patients.
Clinical Presentation
Pulmonologists generally agree that the once dramatic clinical presentation of bronchiectasis (the classic patient with cough and large volumes of purulent and frequently foul-smelling sputum production) is now uncommon. In its place, there is a more insidious form of bronchiectasis in which patients give a typical story of so-called wheezy bronchitis during childhood and chronic purulent rhinosinusitis ; just less than half of those patients with bronchiectasis will also report an antecedent history of pneumonia or another respiratory tract infection. There is then often something of a “remission” in the teenage years, only to be followed, typically after a viral respiratory tract infection, by the more classic symptoms associated with bronchiectasis—namely, chronic cough and mucopurulent sputum production. Many patients also complain of breathlessness, intermittent hemoptysis, pleuritic chest pain, weight loss, and fatigue. The findings on physical examination are nonspecific and include early to midinspiratory crackles and wheezing. Digital clubbing is seen in up to one-quarter of patients with bronchiectasis.
Pathophysiology
Anatomy
With successive generations of bronchial branching, there is a progressive diminution of airway caliber; a lack of tapering is abnormal. The bronchial tree is composed of the large (bronchi) and small (bronchioles) airways. The traditional distinction is based on the presence or absence of cartilage in the wall; airways reinforced by cartilage and typically greater than 1 mm in diameter are termed bronchi , whereas those without cartilage and typically less than 1 mm in diameter are termed bronchioles . Although larger airways are conducive to maximum rate of airflow, a rapid increase in total cross-sectional diameter at the level of the small airways facilitates gas exchange across the alveolar-capillary membrane.
Pathology
The vicious circle hypothesis for the pathogenesis of bronchiectasis, first advocated by Cole and colleagues, is the most widely accepted. The theory is based on the premise that there is initial damage to or disruption of the process of normal mucociliary clearance. This may have been as a result of a viral respiratory infection or a genetic susceptibility, as is the case in patients with cystic fibrosis. The inability to clear mucus permits the “normal” oropharyngeal flora, including Haemophilus influenzae, Pseudomonas aeruginosa, and Streptococcus pneumoniae , to persist within the airways. The organisms lead to further epithelial damage, reducing mucociliary clearance and encouraging further colonization. An inflammatory host response also contributes to further airway damage and dilatation of the airways. With time, there is loss of the elastin layer in the bronchial wall and destruction of cartilage. Some data, based on serial observations by use of computed tomography (CT), suggest that the vicious circle theory may need some refinement, particularly in the pediatric population, because bronchial dilatation diagnosed by high-resolution CT is not always irreversible or progressive.
Three different patterns of bronchiectasis are recognized: cylindrical, varicose, and cystic. In cylindrical bronchiectasis, there is relatively uniform bronchial dilatation, and the implication is that this is perhaps the least severe morphologic type. This should be contrasted with varicose bronchiectasis, in which there are focal narrowings along otherwise dilated airways, and cystic bronchiectasis, which, as the term suggests, indicates grossly dilated airways that have a cystic appearance. Although these morphologic subtypes of bronchiectasis are generally identifiable on high-resolution CT, the clinical utility of making these distinctions is questionable.
Lung Function
The classic physiologic defect in bronchiectasis is airflow obstruction, which in some patients is partially reversible. The obstructive defect has been variably attributed to the presence of coexistent emphysema, airway hyperreactivity or asthma, constrictive bronchiolitis, and expiratory large airway collapse. However, CT studies suggest that areas of decreased attenuation, possibly reflecting constrictive bronchiolitis at the pathologic level, and severity of bronchial wall thickening are the best correlates of obstructive physiology. In some patients with bronchiectasis, there is an associated restrictive ventilatory defect, possibly related to patchy peribronchial fibrosis (presumably consequent on inflammation) and atelectasis.
Manifestations of the Disease
Radiography
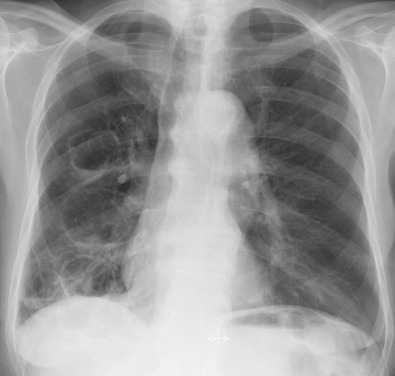
The chest radiograph is generally the first radiologic test requested by the pulmonologist when a diagnosis of bronchiectasis is suspected. However, radiography has low sensitivity and specificity in the diagnosis ( Fig. 57.1 ). However, before the advent of CT and before the conception of the high-resolution CT technique, pulmonologists and radiologists were necessarily reliant on chest radiography, followed in many cases by the then gold standard, but now obsolete, investigation of bronchography, in patients with suspected bronchiectasis ( Fig. 57.2 ). In the frequently quoted study by Gudbjerg, the conclusion was that a mere 10% of patients with bronchiectasis would have a normal chest radiograph. However, although this would have been true at the time, the study was published more than 50 years ago, and as discussed before, the clinical manifestations of bronchiectasis have changed. Moreover, many of the radiographic signs described by Gudbjerg (e.g., linear markings, bronchial wall thickening, and patchy or confluent shadows) are nonspecific. This was clearly illustrated by the later study from the Brompton Hospital that compared chest radiographic diagnoses of bronchiectasis with bronchography and confirmed the highly insensitive nature of chest radiography, also showing considerable disagreement between two experienced pulmonary radiologists simply for confirming the presence or absence of individual features of bronchiectasis.


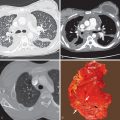
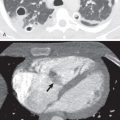
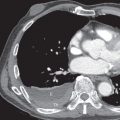
When identified, bronchial dilatation is the key radiographic feature of bronchiectasis. Thus the observer may see poorly defined ring shadows (reflecting the dilated bronchus with peribronchial inflammation when it is seen end-on) or “tram-line” opacities (denoting the nontapering airway), depending on the orientation of the individual airway with respect to the x-ray beam ( Fig. 57.3 ). With more severe disease, obvious thin-walled cysts, with or without air-fluid levels, may be apparent ( Fig. 57.4 ).
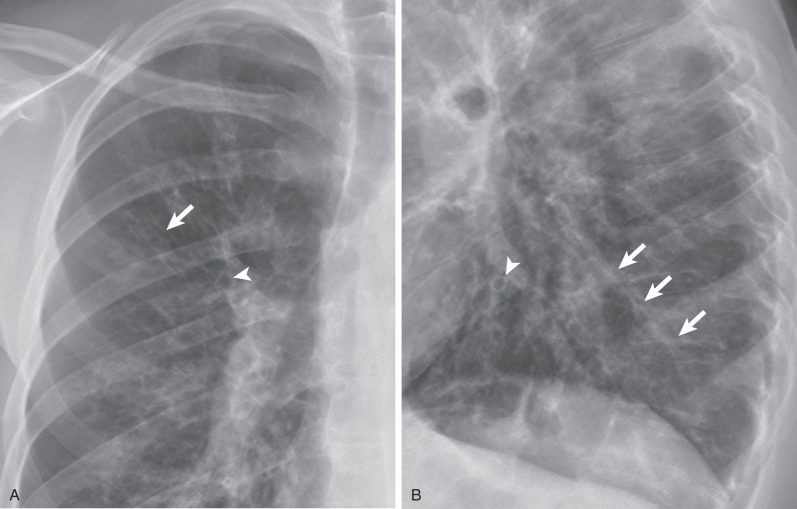
The other recognized features of bronchiectasis on chest radiographs include tubular and branching opacities caused by plugging of dilated airways with mucus, volume loss or hyperexpansion, and foci of subsegmental atelectasis. These are ancillary radiographic signs and must not be regarded as diagnostic of bronchiectasis.
Computed Tomography
In the era before the development of the thin-section CT technique, data on the value of CT in diagnosis of bronchiectasis were disappointing, with sensitivities in the range of 66% to 79%. However, with the realization that spatial resolution could be improved significantly by reducing slice collimation, the reported sensitivities for the CT diagnosis of bronchiectasis improved, with sensitivities approaching 100% using thin-section (1–1.5 mm) imaging.
A factor to be considered in review of the airways on thin-section CT is the window setting because, in the context of a diagnosis of bronchiectasis, this may have a significant influence on the assessment of luminal diameter. In their experimental study, Webb and colleagues showed that for airways surrounded by air, a window centered at −450 Hounsfield units (HUs) provided the most accurate measurement of wall thickness. Indeed, at this window mean, the window width had an imperceptible effect on the evaluation of wall thickness. When the window center was set lower than −450 HUs, wall thickness was overestimated, and the converse was true when the setting was higher.
Computed Tomographic Features of Bronchiectasis
CT signs of bronchiectasis were first described by Naidich and colleagues in 1982, albeit on non–high-resolution CT (10-mm collimation) images. The features identified by these investigators have, with some modifications, largely been validated in subsequent series. Although bronchial dilatation is the key morphologic abnormality, bronchial wall thickening, patchy areas of decreased parenchymal attenuation and vascularity (termed mosaic attenuation ), plugging of large and small airways, volume loss, crowding of airways, and, in some patients with idiopathic disease, thickening of interlobular septa can be seen in association with bronchiectasis.
Bronchial Dilatation.
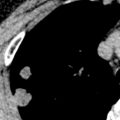
Bronchiectasis is abnormal permanent dilatation of the airways, with or without associated thickening of the walls of the airways. In the normal state the internal diameter of the airway is roughly equal to the transverse diameter of the accompanying pulmonary artery; a bronchoarterial ratio greater than 1 should generally be regarded as abnormal. The CT appearance of bronchiectatic airways depends on the orientation of abnormal bronchi relative to the plane of section. Thus, for airways that are perpendicular to the imaging plane, the radiologist will look for a dilated airway next to a dot representing the normal adjacent pulmonary arterial branch, an appearance that has been likened to a signet ring ( Fig. 57.5 ). In contrast, for bronchi that lie in the plane of section, as in the middle lobe and lingula, the radiologist should look for an absence of normal tapering ( Fig. 57.6 ).


The identification of airway dilatation in patients with severe (varicose or cystic) bronchiectasis does not usually pose a challenge. However, the majority of patients with bronchiectasis now present with mild disease, and the recognition of less severe (cylindrical) bronchial dilatation is certainly problematic. A spurious increase in the bronchoarterial ratio may be seen because the accompanying pulmonary artery can divide before the airway. In this way, the bronchial diameter will appear to be increased compared with the homologous arterial branch. Another consideration is that a bronchoarterial ratio greater than 1 is not always abnormal because it may be seen in up to one-fifth of healthy subjects. Furthermore, it has been shown that there is a normal but progressive increase in the internal diameter of the airways with age, with a bronchoarterial ratio exceeding 1 in just more than 40% of asymptomatic subjects older than 65 years. Finally, altitude may have a bearing on the relationship between airways and their adjacent pulmonary arteries; 50% of healthy subjects had evidence of bronchial dilatation by conventional CT criteria in one study performed in Colorado. Another study confirmed higher bronchoarterial ratios at high altitude compared with sea level. Although the exact mechanisms are not clear, it is conceivable that the increase in bronchoarterial ratio at altitude might be due to vasoconstriction associated with hypoxia, coupled possibly with the phenomenon of hypoxic bronchodilatation.
Kim and colleagues looked for features on CT that would differentiate normal subjects from patients with surgically proven bronchiectasis. This study showed that, in contrast to an increased bronchoarterial ratio and lack of tapering, the visualization of airways within 1 cm of the costal or paravertebral pleura and the identification of airways abutting the mediastinal pleura were more discriminatory, being seen in 81% and 53%, respectively, of the patients with bronchiectasis but in none of the normal subjects.
Ancillary Computed Tomographic Features of Bronchiectasis
Although dilatation of bronchi is the cardinal morphologic manifestation of bronchiectasis, there are a number of ancillary signs on CT. A diagnosis of bronchiectasis cannot be made on the basis of these ancillary CT features in the absence of bronchial dilatation. These additional signs of bronchiectasis are discussed briefly.
Bronchial Wall Thickening.
Thickening of the bronchial wall commonly accompanies bronchiectasis on CT and is likely to reflect an inflammatory component. As has already been mentioned, appreciation of wall thickening is subject to technical factors; window settings that are too narrow can lead to an overestimation of bronchial wall thickness. Interpretation of bronchial wall thickening is also hampered by the lack of consensus on what constitutes normal wall thickness at CT. In an attempt to address this, Remy-Jardin considered bronchi to be thick walled if the wall of any individual airway was at least twice as thick as that of a normal airway. However, the obvious drawback is the assumption that a normal comparable bronchus will be visible; naturally, this cannot be guaranteed when bronchiectasis is severe and widespread. An alternative definition of wall thickening is internal diameter of the airway lumen less than 80% of its external diameter. This definition is useful but only when bronchial dilatation is relatively mild; plainly, if the dilatation is marked (as in cystic bronchiectasis), wall thickening will be underestimated.
Mosaic Attenuation.
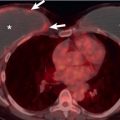
Patchy areas of decreased attenuation, within which there is a reduction in the number or caliber of pulmonary vessels, are not only common but also a functionally important finding on CT in patients with bronchiectasis ( Fig. 57.7 ). The extent of areas of decreased attenuation on expiratory CT does not correlate with indices of gas transfer such as diffusing lung capacity for carbon monoxide, suggesting that areas of mosaic attenuation are due to associated constrictive bronchiolitis.

Regions of air-trapping on expiratory CT tend to be most prevalent in lobes with either extensive or cystic bronchiectasis or in association with large mucous plugs and plugging within the centrilobular bronchioles. However, an additional intriguing finding is the presence of areas of decreased attenuation in some lobes without overt bronchiectasis ( Fig. 57.8 ). On this basis, it has been hypothesized that constrictive bronchiolitis may be the key initial pathologic lesion in bronchiectasis.

Airway Plugging.
Inflammatory mucous secretions may fill the ectatic bronchi, resulting in tubular and nodular opacities representing bronchi seen along their long axis and short axis, respectively. Care must be taken not to miss bronchiectasis in these cases ( Fig. 57.9 ). Mucous secretions within the distal airways and inflammatory thickening of the bronchiolar walls may produce a tree-in-bud pattern, which manifests as Y – or V -shaped opacities or a linear branching structure ( Fig. 57.10 ), depending on the orientation of the airway relative to the imaging plane.


Volume Loss.
Crowding of the airways and volume loss (including, in some patients, complete collapse of a lobe) are recognized features of bronchiectasis that are readily appreciated at CT ( Fig. 57.11 ). It is reasonable to hypothesize that these features of bronchiectasis are a consequence of inflammation and fibrosis around the airways. However, this is not to be confused with the entity of traction bronchiectasis; as the term suggests, this refers to tractional dilatation of airways from adjacent fibrotic interstitial lung diseases (see later).

Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) has a limited role in the diagnosis of bronchiectasis ( Fig. 57.12 ), and the clinical utility of such tests for routine clinical management of patients with bronchiectasis remains debatable.

Imaging Algorithms
Despite its constraints, chest radiography is often the first imaging modality obtained for the evaluation of patients with bronchiectasis. However, given the limited sensitivity and the likelihood that the majority of patients now present with less severe disease, many will require thin-section CT imaging.
Differential Diagnosis
The challenge for radiologists in making a diagnosis of bronchiectasis is twofold: first, in ensuring that the diagnosis is not being overcalled, because of one of the potential mimics, and second, in trying to determine a possible cause of bronchiectasis.
The distinction between true bronchiectasis and lung diseases characterized by cysts, such as pulmonary Langerhans cell histiocytosis and lymphangioleiomyomatosis, may be rendered difficult, particularly if they are severe. In practice, the experienced radiologist will take care to check adjacent sections; plainly, dilated airways will demonstrate continuity, whereas in diseases characterized by intrapulmonary cysts, such continuity will not be seen. Finally, mention must be made again of the term traction bronchiectasis, which is occasionally a source of confusion to physicians on radiologic reports ( Fig. 57.13 ). It seems likely that such dilatation of airways, a relatively common finding in patients with interstitial fibrosis, is due to the radial traction on the airways by thickened alveolar attachments in areas of fibrosis. The important clinical point for the pulmonologist, of course, is that patients with this finding do not present with the clinical picture of patients with true bronchiectasis.

The craniocaudal and axial distribution of abnormalities may help narrow the differential diagnosis of a particular case of bronchiectasis. Bronchiectasis in cystic fibrosis is more common in the upper zones. Similarly, allergic bronchopulmonary aspergillosis is generally regarded as a central and upper zonal disease, whereas bronchiectasis that is related to aspiration, immunodeficiency, α1-antitrypsin deficiency, or iatrogenic causes appears to have a predilection for the lower zones. Finally, it might reasonably be expected that bronchiectasis occurring after pneumonia would be relatively localized, occurring in the area affected by the pneumonia. However, consideration of both imaging findings and clinical scenario may lead to a more accurate diagnosis than imaging alone.
Specific Causes of Bronchiectasis
Cystic Fibrosis
Etiology, Prevalence, and Epidemiology
Cystic fibrosis (CF) is an autosomal-recessive hereditary disease. It is the most common lethal genetically transmitted disease in white individuals; the estimated incidence is about 1 per 2000 to 3500 live births. CF is uncommon in nonwhites. There is no sex preponderance. The diagnosis is made before age 5 years in about 80% of cases, during adolescence in 10%, and during adulthood in 10% of cases. The fundamental abnormality consists of the production of abnormal secretions from exocrine glands, such as the salivary and sweat glands, and the pancreas, large bowel, and tracheobronchial tree. The major clinical manifestations are obstructive pulmonary disease, which is found in varying degrees of severity in almost all patients, and pancreatic insufficiency, present in 80% to 90%. CF is the most common cause of pulmonary insufficiency in the first 3 decades of life.
Clinical Presentation
The pulmonary manifestations of CF include recurrent respiratory infections associated with productive cough, wheezing, and dyspnea. The infection is predominantly caused by bacteria, such as Pseudomonas aeruginosa , Staphylococcus aureus , and Haemophilus influenzae , although viruses, mycoplasma, and fungi are occasionally responsible. The presence of Burkholderia cepacia is usually associated with advanced disease. Common complications include hemoptysis and pneumothorax. Most patients eventually progress to respiratory insufficiency accompanied by pulmonary arterial hypertension and cor pulmonale.
Pathophysiology
Pathology
The earliest pathologic lesion of CF is obstruction of bronchioles and small bronchi by abnormal mucus, which is followed by airway inflammation and infection. Pulmonary disease in patients with CF typically progresses from bronchiolitis and bronchitis to bronchiectasis owing to chronic infection and airway obstruction by the inspissated mucus. Focal emphysema and bullae also can be seen. Histologic examination typically reveals chronic inflammation and fibrosis of the bronchial wall, partial or complete luminal occlusion by purulent material, focal epithelial ulceration, and cartilage destruction.
Lung Function
Pulmonary function tests show progressive airway obstruction and air-trapping, with decreased forced expiratory volume at 1 second, increased vital capacity, and increased residual volume.
Manifestations of the Disease
Radiography
The earliest manifestations of CF consist of round or poorly defined linear opacities measuring 3 to 5 mm in diameter that are located within 2 to 3 cm of the pleura. Less common early manifestations include thickened bronchial walls without bronchial dilation, usually seen as ring shadows, and mild hyperinflation. Progression of disease is characterized by increases in bronchial diameter, bronchial wall thickness, lung volume, and number and size of peripheral nodular opacities and by the development of mucoid impaction and focal areas of consolidation ( Figs. 57.14 and 57.15 ). Bronchiectasis, bronchial wall thickening, and mucous plugging are particularly frequent and are evident in almost all adult patients. The chest radiographic abnormalities have been incorporated into numerous semiquantitative scoring schemes that are believed to be of value in predicting prognosis and directing therapy.


Recurrent foci of consolidation occur in most patients, and lobar or segmental atelectasis occurs in many. Hilar enlargement may be due to lymph node enlargement or dilation of the central pulmonary arteries secondary to pulmonary arterial hypertension (see Fig. 57.15 ). Pneumothorax occurs in 3% to 19% of patients.
Computed Tomography
The main manifestation of CF is bronchiectasis, which is present in virtually all adult patients ( Fig. 57.16 ; see Fig. 57.14 ). Bronchiectasis usually involves all lobes but tends to be most severe in the upper lobes. The bronchiectasis may be cylindrical, varicose, or cystic. Other common findings are bronchial wall thickening, peribronchial interstitial thickening, and mucous plugging. Consolidation or atelectasis can be seen in 80% of cases. Cystic or bullous lung lesions also can be seen and typically predominate in the subpleural regions of the upper lobes (see Fig. 57.16 ).

Branching or nodular centrilobular opacities (tree-in-bud pattern) are frequently present and may be an early sign of disease. They reflect the presence of bronchiolar dilation with associated mucous impaction, infection, or peribronchiolar inflammation. Focal areas of decreased attenuation and vascularity on inspiratory CT and air-trapping on expiratory CT are common.
Hilar or mediastinal lymph node enlargement and pleural abnormalities may be present, largely reflecting chronic infection. Pulmonary artery dilation resulting from pulmonary hypertension is common in patients who have long-standing disease.
Magnetic Resonance Imaging
MRI may be used as an alternative tool to assess parenchymal and functional abnormalities and to monitor treatment in patients with CF. Although MRI has lower spatial resolution than CT and plays a limited role in the assessment of airway disease, it has the advantage of no radiation exposure, an important consideration in these young patients. MRI may be useful in the follow-up of patients with CF ( Fig. 57.17 ) and could be used to screen for early liver disease, given the increased life expectancy of CF patients in recent years.

Imaging Algorithms
The chest radiograph is the main imaging modality used in the initial evaluation and follow-up of patients with CF. High-resolution CT can show abnormalities in patients who have early CF and normal chest radiographs. In one study of 38 patients who had mild CF with normal pulmonary function, chest radiographs were normal in 17 (45%), showed mild bronchial wall thickening in 17, and showed mild bronchiectasis in 4 (10%). On high-resolution CT in this group, bronchiectasis was present in 77% of all patients and in 65% of patients with normal chest radiographs; only 3 patients had a normal high-resolution CT scan.
When performing repeated high-resolution CT scans in young patients, the risk of radiation has to be considered and the radiation dose minimized. Axial high-resolution chest CT with interspace gaps can be performed at a much lower dose than volumetric scans. When volumetric CT is required, it is recommended that this be performed with low milliamperage (40 mA) and low kilovoltage.
Differential Diagnosis
The radiologic findings of CF may mimic those of other causes of bronchiectasis, such as allergic bronchopulmonary aspergillosis and postinfectious bronchiectasis. The most characteristic features of bronchiectasis in CF are bilateral symmetric distribution and upper lobe predominance. In one study, bronchiectasis in CF involved mainly the upper lobes in 79% of patients and was usually bilateral and fairly symmetric, whereas the bronchiectasis in the other conditions, such as prior tuberculosis, was most frequently unilateral or asymmetric.
Although the diagnosis of CF may be suggested by a positive family history, persistent respiratory disease, clinical evidence of pancreatic insufficiency, or fatty (or less common cystic) replacement of the pancreas on imaging, confirmation requires a positive sweat test or identification of two abnormal copies of the CF gene. Incorporation of molecular biologic techniques into rapid, cost-efficient, and specific diagnostic tests for most CF genotypes is now possible by using the multiplex polymerase chain reaction.
Synopsis of Treatment Options
Therapy typically includes antibiotics, physiotherapy, and replacement of pancreatic enzymes. Once a disease with a life expectancy confined to the adolescent years, now most patients reach adulthood. The median survival of CF patients has increased significantly over the past few decades to almost 41 years in the United States and approximately 51 years in Canada.
- •
The estimated incidence is 1 per 2000–3500 live births in whites; it is uncommon in nonwhites.
- •
The main clinical manifestations are obstructive pulmonary disease and pancreatic insufficiency.
- •
Radiographic findings include bronchial wall thickening, bronchiectasis, hyperinflation, and areas of consolidation or atelectasis.
- •
The main CT manifestation is upper lobe–predominant bronchiectasis.
- •
Other common findings are bronchial wall thickening, peribronchial interstitial thickening, mucous plugging, branching or nodular centrilobular opacities (tree-in-bud pattern), focal areas of decreased attenuation and vascularity on inspiratory CT, and expiratory air-trapping.
- •
Judicious use of CT scans and certain CT dose-reduction techniques is important in minimizing radiation exposure in CF patients.
Primary Ciliary Dyskinesia (Dyskinetic Cilia Syndrome)
Etiology, Prevalence, and Epidemiology
The term primary ciliary dyskinesia refers to a group of autosomal-recessive disorders associated with defective ciliary structure and function, also known as dyskinetic cilia syndrome. The structurally abnormal cilia move ineffectively and predispose to sinusitis, recurrent pulmonary infections, and bronchiectasis. Situs inversus totalis or, less commonly, heterotaxy syndrome is seen in about 50% and 12% of patients, respectively. Men have reduced fertility as a result of decreased motility of the spermatozoa. Although primary ciliary dyskinesia has an autosomal-recessive pattern of inheritance, the variety of ultrastructural defects associated with the clinical syndrome suggests considerable genetic heterogeneity. The incidence of the syndrome in white individuals is estimated to be 1 in 12,500 to 40,000; a higher prevalence has been reported in Japan.
Clinical Presentation
The age of presentation ranges from 4 months to 51 years. The clinical manifestations include infertility, chronic rhinitis, sinusitis, otitis, and recurrent lower respiratory tract infections. The most common bacterial organism found in sputum is H. influenzae , but Pseudomonas isolates also are common. Half of patients have Kartagener syndrome, defined by the triad of situs inversus totalis, bronchiectasis, and either nasal polyps or recurrent sinusitis. Morbidity in primary ciliary dyskinesia is predominantly related to chronic suppurative airway disease secondary to chronic infection.
Pathophysiology
Pathology
Ultrastructural abnormalities in primary ciliary dyskinesia seen on electron microscopy include a lack of outer dynein arms, absent or short radial spokes, absent or defective inner dynein arms, absent or disoriented central microtubules, and transposition of peripheral microtubules. A combination of structural defects is often present.
Lung Function
Many patients develop mild to moderate airflow obstruction.
Manifestations of the Disease
Radiography
Radiographically, abnormalities progress from bronchial wall thickening to bronchiectasis, hyperinflation, segmental atelectasis, and consolidation. In one study of 30 patients ranging from newborn to 26 years old, radiographic abnormalities were evident in all patients. Findings usually have a lower zone distribution, but radiologic features may resemble the features of bronchiectasis from a variety of other causes; when present, the combination of lower zone–predominant bronchiectasis and situs inversus is nearly pathognomonic for the diagnosis ( Fig. 57.18 ).