CHAPTER 15 Congenital Cholesteatoma
Etiology
Congenital cholesteatomas are distinct from acquired cholesteatomas of the middle ear. Previously thought to be rare, they are being more frequently diagnosed in young children. Congenital cholesteatomas are known to develop in four temporal bone sites: the petrous apex, the middle ear, the mastoid, and the external auditory canal. Some can affect both the middle ear and the mastoid. We limit our discussion here to middle ear lesions, as other locations are discussed elsewhere in this book.
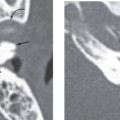
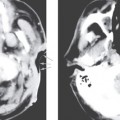
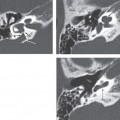
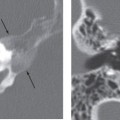
The current established criteria for diagnosis for a congenital cholesteatoma was described in 1986 by Levenson et al, who modified the original criteria of Derlacki and Clemis from 1965. The criteria now include a sac medial to an intact tympanic membrane in a patient who has never had a previous tympanic membrane perforation, otorrhea, or prior otologic surgery. Prior otitis media does not completely exclude the diagnosis. These lesions are most often unilateral, though bilateral lesions have been reported.
Several mechanisms have been proposed to explain the pathogenesis of congenital cholesteatoma, including persistent epidermoid cell rest, ingrowth of meatal epidermis, and reflux of amniotic fluid. Of these, the favored theory of pathogenesis appears to be the persistence and continued growth of the epidermoid formation that normally exists in the middle ear during fetal life and usually disappears after 33 weeks of gestation. The theory also explains the more common anterior-superior quadrant location of these lesions.
Clinical Features
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


