Abstract
A congenital cystic adenomatous malformation is a developmental lung malformation consisting of abnormal hamartomatous or dysplastic lung tissue and bronchoalveolar structures. The diagnosis can be made during fetal life through an ultrasound examination of the fetal thorax that identifies a hyperechoic cystic or solid-cystic lung mass with blood supply originating in the pulmonary artery. The prognosis is usually good but depends on the lung mass size, the degree of pulmonary compression, and the presence of fetal fluid effusions or hydrops. In those cases with poor prognosis (at risk of perinatal death), different fetal interventions have demonstrated potential benefits in improving the survival rate. However, such procedures are not curative, and therefore all cases require postnatal surgical resection of the lung mass by toracoscopy or toracotomy.
Keywords
congenital cystic adenomatoid malformation (CCAM), congenital lung lesions, hyperechoic lung, fetal cystic lung lesion, fetal intervention
Introduction
Congenital cystic adenomatous malformations (CCAMs) of the lung represent the most common lung defect diagnosed prenatally.
CCAMs are intrapulmonary lesions with a typical hyperechoic appearance on ultrasound (US), with or without cystic components. Both sides of the lung, both sexes, and all races are equally affected. Most fetuses with CCAM are detected prenatally and have a good outcome, but appropriate identification and ongoing surveillance are required because of the unpredictability of growth patterns for CCAM masses.
Disorder
Definition
CCAM is a developmental, nonhereditary, usually mixed (solid and cystic) lung mass consisting of abnormal hamartomatous or dysplastic lung tissue and bronchoalveolar structures thought to result from abnormal branching of the immature bronchioles during early stages of lung morphogenesis.
Prevalence and Epidemiology
CCAM is the most common fetal hyperechogenic lung lesion and accounts for 50% to 75% of detected fetal lung abnormalities. These lesions may display dramatic changes during pregnancy, with spontaneous regression and total resolution in more than half of cases. Thus, postnatal studies probably underestimate the real incidence of lung lesions, with a commonly quoted incidence of 1 : 25,000 to 1 : 35,000. In prenatal studies performed in nonreferred populations, an incidence of 1 : 4000 to 1 : 6000 has been reported. Prenatal diagnosis has dramatically increased as a result of improvement of US equipment over time, so the precise prevalence of prenatal CCAM is still unknown and might change in the future.
Etiology and Pathophysiology
CCAM is characterized by lack of normal alveoli and originates from a dysplastic overgrowth and cystic dilatation of terminal bronchioles with various types of epithelial lining.
CCAM may have a mainly solid, mainly cystic, or mixed appearance. Stocker et al. described three types of lung lesions based on cyst diameter, which probably represent differences in the origin of the dysplastic lesion in the bronchial tree levels.
- •
Type I macrocystic CCAMs are characterized by single or multiple cysts greater than 2 cm in diameter, lined by ciliated pseudostratified columnar epithelium. These represent nearly 50% of CCAM cases in postnatal series. They frequently cause mediastinal compression, are rarely associated with other anomalies, and generally have a good prognosis.
- •
Type II lesions account for up to 40% of CCAMs, are single or multiple cysts less than 2 cm in diameter, and are lined with mixed ciliary, columnar, and cuboidal epithelium, with a thin underlying fibromuscular layer.
- •
Type III lesions are microcystic, predominantly solid lesions, with small cysts (<0.5 cm). These lesions represent 10% of CCAM lesions and are histologically composed of alveoluslike structures lined by ciliated cuboidal epithelium separated by microscopic masses lined with nonciliated cuboidal epithelium.
Stocker et al. later expanded his classification to include another two types. Type 0, acinar dysplasia or agenesis, is very rare, involves all the lung lobes, and is incompatible with life. Type IV CCAM lesions are represented by large, thin-walled peripheral cysts caused by hamartomatous malformation of the distal acinus.
Another, more clinically appropriate classification, based on US appearance, was introduced by Adzick et al., who divided prenatally diagnosed lung lesions into two groups: macrocystic (type I), with cysts greater than 5 mm, which is seen on US as a cystic mass, and microcystic (type II) with cysts smaller than 5 mm, which appear as solid echogenic lesions on US. The presence or absence of cysts is important because this determines options for therapy in cases of fetal hydrops.
Manifestations of Disease
Clinical Presentation
A CCAM appears as a hyperechoic solid or cystic thoracic mass. Cysts may be single or multiple, small or occupying the entire volume of the mass. CCAM is usually unilateral and unilobar, with a slight predilection for the lower lobes of the lung. The mass is usually detected in the second trimester, commonly showing slight growth at the beginning. In half of cases, there is apparent prenatal resolution of the hyperechoic lesion, usually by around 32 weeks’ gestation. Large tumors are associated with mediastinal shift with displacement of the heart to the contralateral part of the thorax, flattening of the diaphragm, esophageal compression resulting in polyhydramnios, and direct cardiac compression and obstruction of venous return resulting in rare cases in hydrops fetalis.
The risk of chromosomal abnormalities is not increased significantly in isolated CCAM. Associated anomalies are detected in 8%–12% of cases. Commonly reported associated anomalies are renal defects, congenital diaphragmatic hernia, tracheoesophageal fistula, and congenital heart defects.
The prognosis of prenatally diagnosed CCAM lesions depends on the size of the lesion and the degree of pulmonary hypoplasia, existence of associated anomalies, and development of fetal hydrops. Evaluation of the size of the lung mass by means of the c ystic adenomatoid malformation v olume r atio (CVR = length × height × width × 0.52 divided by the head circumference) is useful for predicting the development of hydrops. A CVR greater than 1.6 presents a 75% risk to develop hydrops, whereas a CVR less than 1.5 has a 3% risk of hydrops.
Imaging Technique and Findings
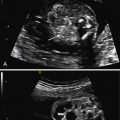
Ultrasound.
An echogenic cystic lung mass. The location, size, and existence of discrete lung cysts and blood supply must be evaluated using conventional spectral, color, or power Doppler US. Type I lesions are predominantly cystic masses with large cysts ( Fig. 2.1 ), type II lesions are predominantly solid masses with small cysts ( Fig. 2.2 ), and type III lesions are solid, homogeneous, hyperechoic masses ( Fig. 2.3 ).